Article Text

Abstract
Perivascular epithelioid tumours (PEComas) of the gynaecological tract are rare tumours which were first recognised and diagnosed within the last 20 years. They represent a unique diagnostic challenge with regard to their accurate and reproducible distinction from more common entities such as smooth muscle tumours of the uterine corpus. In this review article, we trace the development of the concept of the PEComa tumour family, highlight what is known about extra-gynaecological tract PEComa at an immunohistochemical, molecular and therapeutic level and then present a summary of all reported cases of gynaecological tract PEComa to date. In the summary, we highlight rare subtypes of gynaecological tract PEComa and compare the performances of extant prognostic classification systems for malignancy in these tumours.
- GYNAECOLOGICAL PATHOLOGY
- NEOPLASMS
- HISTOPATHOLOGY
- DIAGNOSIS
- UTERUS
Statistics from Altmetric.com
Introduction: PEComa
Demonstration of the expression of the melanocytic marker HMB-45 in angiomyolipoma (AML)1 ,2 and clear cell ‘sugar’ tumours (CCSTs) of lung3 led to the concept of PEComa as a family of tumours occurring at many sites and characterised by the presence of an epithelioid cell of mixed myomelanocytic immunophenotype. This family included previously recognised entities such as AML, CCST, lymphangioleiomyomatosis (LAM) and clear cell myomelanocytic tumour of the falciform ligament/ligamentum teres. Subsequently, sporadic HMB-45-positive clear cell tumours of other sites such as pancreas4 and uterus5 were described.
The cell of origin of PEComas has not been unequivocally established. Early reports speculated tumour origin from vessel walls6 or from a ‘peculiar muscle cell’ based on morphology and expression of myomelanocytic markers.3 Embryological and in vitro studies have provided evidence for origin from a neural-crest stem cell that is capable of myoid and melanocytic differentiation during embryological development and has also been demonstrated in the context of tissue repair.7–11
Tuberous sclerosis complex (TSC) is characterised by the development of tumours at various sites, including brain, heart and kidney. Genetically, TSC is associated with mutations in TSC1 or TSC2 (located on 9q 34 and 16p13 respectively), leading to impaired production of the proteins hamartin and tuberin, respectively. TSC1 and TSC2 interact as heterodimers that inhibit the mechanistic target of rapmycin (mTOR) pathway; their inactivation leads to increased cell growth and proliferation.12 The prototypical PEComa associated with TSC is renal AML. While ∼80% of patients with TSC have AML, <50% of all renal AMLs and <10% of extrarenal AML occur in patients with TSC.13 The majority of reported cases of PEComatosis (widespread multifocal macroscopic and microscopic nodules of PEComa cells involving multiple sites in the gynaecological tract and pelvis) have occurred in TSC patients,14–18 while the uterus of a single TSC patient harboured a subserosal AML, a sclerosing PEComa of lower uterine segment and diffuse LAM of the uterine corpus.19 Thus, TSC-associated loss of function of TSC1/TSC2 can lead to a phenotypic spectrum (classical AML-like, classical PEComa, sclerosing PEComa, LAM) that is also recognised in sporadic PEComas.
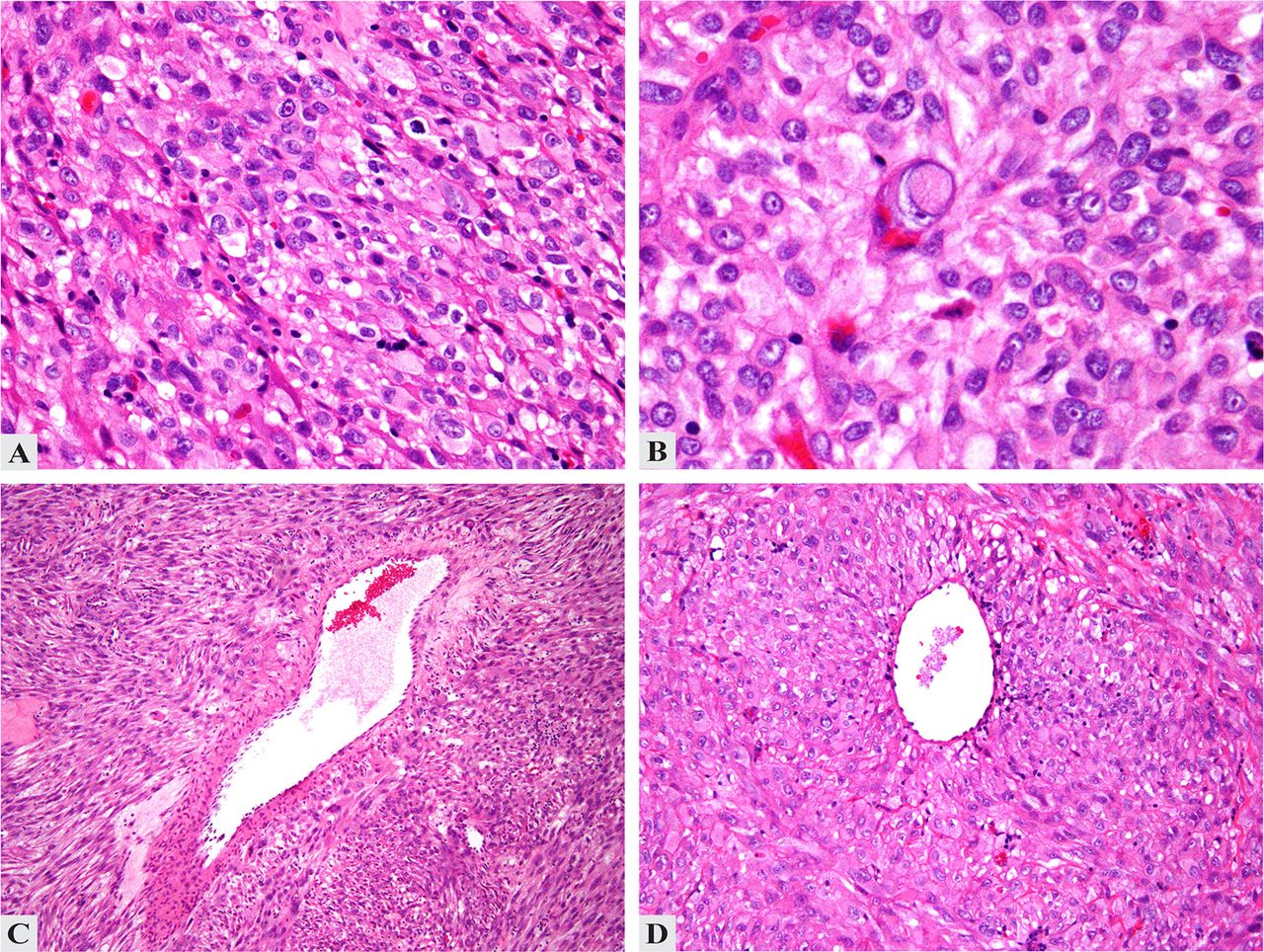
Histologically, PEComa is characterised by the presence of predominantly epithelioid cells with clear, granular or eosinophilic cytoplasm, arranged in nests or sheets, with little intervening stroma (figure 1A–D). Many PEComas have a low mitotic rate of 0–1 per 50 high power fields (HPFs). Areas of necrosis may be seen (figure 2A). PEComas demonstrate varying levels of nuclear pleomorphism, including multinucleate giant cells (figure 2B) and ‘spider cells’, analogous to those seen in cardiac rhabdomyoma.20 Other features include macronucleoli (figures 1C and 2B) and intranuclear pseudoinclusions (figure 1C).20 While most reported PEComas have been epithelioid, they show a cytological spectrum from purely spindled to purely epithelioid (and combinations of the two). Rare features include sex-cord-like features21 and prolactin secretion.22 ,23

Histopathological features of uterine perivascular epithelioid tumour. (A) Sheets of epithelioid cells without intervening stroma. (B) Cells with abundant eosinophilic, fibrillary cytoplasm, prominent nucleoli and occasional nuclear pseudoinclusions. (C and D) Perivascular cuffing by tumour cells.

{kind=link}
{kind=link}
Malignant perivascular epithelioid tumour of uterus. (A) Pleomorphic tumour with necrosis (upper left). (B) Multinucleate tumour cells with macronucleoli.
Sclerosing PEComa occurs predominantly in the retroperitoneum of women and rarely in the uterus and pelvis. They show cords of epithelioid cells within densely sclerotic stroma. Areas of intimate association between tumour cells and blood vessels are often identified.24
PEComas are defined by the immunohistochemical (IHC) coexpression of myoid markers (smooth muscle actin (SMA), desmin, caldesmon) and melanocytic markers (HMB-45, Melan-A, microphthalmia-associated transcription factor (MiTF)). Expression varies with morphology: tumours with predominant spindle cell morphology show strong expression of muscle markers and limited expression of melanocytic markers; predominantly epithelioid tumours may strongly express melanocytic markers with limited muscle marker expression. Recently, cathepsin K, a transcriptional target of the MiTF family, emerged as a sensitive marker for PEComa.25–27 However, cathepsin K is not specific for PEComa and is commonly positive in melanoma, alveolar soft part sarcoma and mesenchymal tumours, including leiomyosarcoma (LMS).28
Renal and extrarenal AMLs/PEComas exhibit true melanocytic differentiation in the form of melanosomes at various stages of development on electron microscopy29 ,30 and positive Masson-Fontana staining. Grossly and microscopically pigmented examples of PEComa have been reported.31–34
Limited genomic studies have suggested that loss of heterozygosity (LOH) at the TSC2 locus may play an important role in sporadic PEComa tumourigenesis, similar to its role in TSC-related and sporadic renal AML35–37 and sporadic pulmonary LAM.38 Comparative genomic hybridisation on nine PEComas (including one uterine case) showed multiple chromosomal imbalances, with 16p loss in six cases and X-chromosomal gains in six cases.39 Malinowska et al40 demonstrated loss of IHC expression of tuberin in four PEComas and LOH or allelic loss of at least one TSC2 microsatellite marker in two of those four cases. Kenerson et al41 reported IHC evidence of mTOR pathway activation (increased levels of phospho-p70S6K, reduced levels of phospho-AKT and loss of tuberin expression) in sporadic AMLs and extrarenal PEComas. Based on these limited studies, several clinical trials of mTOR pathway inhibitors in malignant PEComa have been initiated.
PEComa of the gynaecological tract
Uterine corpus
To date, 78 cases of uterine corpus PEComas have been reported in the English-language literature (table 1).5 ,16–20 ,24 ,39 ,42–72 Tumour morphology was described in 74 cases (table 1). A summary of the reported IHC findings is presented in table 2. A mixed myomelanocytic phenotype, with positivity for at least one melanocytic and one muscle marker, was confirmed in 66 of 73 (90%) cases. The remaining tumours44 ,45 ,48 ,71 (including three transcription factor E3 (TFE3)-translocation-associated PEComas71) were HMB-45-positive but negative for multiple muscle markers.71 On ultrastructural examination, seven of 11 (64%) tumours5 ,42 ,48 ,52 ,59 ,70 showed evidence of melanocytic differentiation (presence of premelanosomes or melanosomes).
Summary of clinicopathological features of PEComas of the gynaecological tract
Expression of myoid and melanocytic immunohistochemical markers in uterine corpus PEComas
Reported chromosomal copy number alterations include trisomy X (n=1)61 and multiple gains and losses including 16p11.1-p13.3, which contains the TSC2 locus (n=1).39 Conversely, no evidence of LOH at the TSC1 or TSC2 loci was identified in another PEComa.52
Twelve patients had evidence of tumour metastasis at the time of diagnosis.20 ,42 ,44 ,54 ,55 ,58 ,60 ,69 Median survival for those who died of disease was 20 months (table 1).
Cervix
Eleven cervical PEComas have been reported.28 ,33 ,53 ,73–79 One tumour demonstrated X polysomy and rearrangement of TFE3,33 whereas another tumour showed biallelic somatic deletion of TSC1.78 One patient (11%) recurred locally within 4 months, but had no evidence of disease at 19 months post diagnosis (table 1).79
Vagina
Seven vaginal PEComas have been reported20 ,53 ,67 ,71 ,80–82 (table 1). Three patients were aged <18 years.80–82
Adnexa
Six adnexal PEComas have been reported to date.20 ,32 ,83–85 (table 1). A mixed myomelanocytic immunophenotype was demonstrated in five cases.20 ,84 ,85 TFE3 rearrangement was identified in one tumour.32 A 63-year-old patient who died of disease at 4 months had a 15 cm sclerosing PEComa.84 While the presence of severe atypia was noted, the presence of necrosis and mitotic rate was not recorded.
Broad ligament
Five PEComas of the broad ligament have been reported27 ,31 ,34 ,53 ,86 (table 1). A mixed myomelanocytic immunophenotype was demonstrated in all four tested tumours.27 ,31 ,34 ,86 Ultrastructural studies in a single tumour showed premelanosomes, consistent with melanocytic differentiation.31
Subtypes of PEComa in the gynaecological tract
PEComatosis
Six cases of PEComatosis have been described in the gynaecological tract.16–18 ,66 ,76 ,88 Median patient age was 43.5 (range 29–70) years. Four (67%) patients had TSC.16–18 ,66 The dominant tumour mass was located in the uterine corpus (n=4),16–18 ,66 vaginal remnant post hysterectomy (n=1)88 and cervix (n=1).76 Other sites involved by PEComatosis included ovary (n=4), lymph nodes, broad ligament, omentum, peritoneum and small bowel wall. Dominant tumour size ranged from 0.8 to 6 cm (mean 2.9 cm).
The dominant tumours showed mixed epithelioid and spindled morphology (three of four tumours), while one tumour was wholly spindled.16 Necrosis was present in one of five cases,18 and mitoses ranged from ‘rare’ and less than or one to 20 per 50 HPF. Atypia was noted in four tumours (moderate in three). HMB-45 was positive in five of five cases (‘strongly’ positive in three of four where extent/intensity was reported), while Melan-A was strongly positive in three of three cases. SMA was positive in six of six cases (three of three strongly positive), while desmin was positive in three of four cases. No ultrastructural studies have been reported in PEComatosis. One case showed balanced chromosomal studies.76
Follow-up information was reported in four of six cases;16 ,66 ,76 ,88 three of four patients had no evidence of disease after 12 to 168 months of follow-up. One patient was alive with disease at 12 months.88
Sclerosing PEComa
Reported cases have involved the uterine corpus (n=9),16 ,19 ,24 ,43 ,64 adnexa (n=2)84 ,85 and broad ligament (n=1).27 Two patients had TSC,16 ,19 and one patient had PEComatosis.16 Median patient age (n=11) at diagnosis was 46 (range 29–63) years. Mean tumour size (n=10) was 3.75 cm (range 0.8–15 cm). Four tumours were purely epithelioid, one was spindled and one was mixed epithelioid-spindled. No necrosis or atypia was identified (n=6) and no mitotic activity was seen (n=5).
IHC for HMB-45 was positive in nine of nine tumours (‘strong’ or ‘diffuse’ positivity in six of eight). Melan-A was negative in five of six cases while MiTF was negative in five of five cases. SMA was strongly and/or diffusely positive in eight of eight tested tumours, while seven of seven tested tumours were positive for desmin (six of seven ‘strong’ or ‘diffuse’). No ultrastructural evidence of melanocytic differentiation was seen in a single tumour.43
Nine patients had no disease recurrence with a median follow-up of 19.5 months (range 12–168 months).16 ,19 ,24 ,43 ,64 ,84 One patient died of her disease 4 months after diagnosis.84
TFE3 translocation-associated PEComa
TFE3 is a member of the MiTF. Translocations involving the TFE3 locus at Xp11.2 have been reported in epithelioid clear cell tumours such as alveolar soft part sarcoma and Xp11.2 translocation-associated renal cell carcinoma. In recent years, 18 cases of TFE3 translocation-associated PEComa have been reported in kidney,25 ,89 bladder,90 colon,91 ,92 pelvic soft tissue,25 ovary,32 vagina71 and uterus.58 ,71 ,91 These tumours appear to be characterised by predominantly epithelioid, clear cell morphology without pleomorphism and IHC positivity for HMB-45, TFE3 and Cathepsin K, and negativity for MiTF,91 ,92 SMA and desmin.25 ,32 ,71 ,91 ,92 This suggests that TFE3-translocation associated PEComa may represent a distinct subgroup within the PEComa family.71 Activation of the mTOR pathway may not necessarily play a role in these tumours, which has implications with respect to patient entry into clinical trials of mTOR pathway inhibitors.40
PEComas associated with TFE3 translocations are immunoreactive for TFE3, but the converse is not necessarily true; for example, three gynaecological tract PEComas which were TFE3-positive on IHC were negative for TFE3 translocation on fluorescent in-situ hybridisation (FISH).71 Since TFE3 is ubiquitously expressed at a low level in many cell types, the use of sensitive IHC techniques may yield positive IHC results in tumours which lack TFE3 translocations.91 Therefore, weak IHC staining for TFE3 on should be interpreted with caution. At present, our approach to TFE3-immunoreactive tumours is to perform FISH to confirm the presence of the translocation.
Assessment of malignancy in uterine PEComa
The prognostic classification system of Folpe et al53, based on retrospective analysis of 26 PEComas of multiple sites, divided PEComas into benign, uncertain malignant potential (UMP) and malignant groups based on histological criteria (table 3). Subsequently, some deficiencies in this system became apparent. While the categorisation of cases with no worrisome features (benign) or two or more worrisome features (malignant) is straightforward, it is unclear how to categorise those PEComas with a single worrisome feature such as elevated mitotic count, necrosis or infiltrative growth pattern.
Folpe criteria for prognosis in PEComa (from Folpe et al53)
Recently, Schoolmeester et al20 applied the Folpe criteria to 16 gynaecological tract PEComas and proposed a revised system (table 4) which set a higher threshold for malignancy (four or more worrisome features) and yielded greater specificity and positive and negative predictive values for subsequent malignant behaviour without sacrificing sensitivity. In addition, they combined the benign and UMP categories into one group, in which no malignant behaviour was observed during the limited follow-up period of the study.
Schoolmeester criteria for prognosis in PEComa (from Schoolmeester et al20)
We extracted all reported histological and outcome data from 78 reported uterine PEComas in order to compare the Folpe and Schoolmeester systems. In an attempt to rectify the deficiencies in the Folpe system outlined above, we tested a modification of the Folpe criteria (modified-Folpe), wherein tumours with a single ‘worrisome’ feature such as maximum dimension of ≥5 cm, infiltrative edge, lymphovascular invasion (LVI) or mitotic count greater than one per 50 HPF are considered benign (table 5). It was not possible to define an upper limit to tumour size or mitotic count due to the small number of reported tumours with a single worrisome feature, but pending data from additional cases, clinicians may use their judgment and move the tumour to the UMP category if they consider the mitotic count or tumour size worryingly high (criteria arbitrarily used in this analysis were tumour size >10 cm or mitoses greater than three per 50 HPF). Tumours with isolated marked atypia, maximum dimension >10 cm or mitotic count greater than three per 50 HPF in the absence of other worrisome criteria should be considered UMP tumours due to the lack of available data. Since necrosis was the single worrisome feature in only one case, in which the tumour was associated with a lymph node metastasis,55 we have placed it in the malignant category.
Summary of modified-Folpe criteria
For our analysis, it was necessary to discard ‘cellularity’ as a criterion as it is poorly defined and is not assessed in most reports. Furthermore, features such as ‘infiltrative edge’ and LVI were not explicitly reported in many papers. We assumed that LVI was not identified if it was not explicitly stated as being present. Tumours in which the nature of the advancing edge was not reported were considered non-assessable using the Folpe and Schoolmeester criteria (unless the tumour reached thresholds for malignancy based on other worrisome features).
Using both Folpe and modified-Folpe criteria, no benign or UMP tumour behaved in a malignant fashion, although the modified-Folpe criteria allowed more tumours to be categorised and recognised as benign (table 6). The Schoolmeester criteria showed superior specificity and positive predictive value (with all 16 tumours categorised as ‘malignant’ showing metastasis or local recurrence) than the Folpe and modified-Folpe systems, but seven of 47 tumours (15%) classified as benign/UMP behaved in a malignant fashion (table 6).
Comparative analysis of PEComa classifications
The Folpe and modified-Folpe criteria show greater sensitivities and negative predictive values than the Schoolmeester criteria. However, tumours meeting the more stringent Schoolmeester definition of malignancy frequently recurred early. Until data from larger numbers of tumours are available, the modified-Folpe criteria may be used first to categorise tumours, with subsequent application of the Schoolmeester criteria to help identify malignant tumours at high risk of early recurrence.
Challenges in diagnosis of gynaecological tract PEComa: the PEComa/smooth muscle tumour morphological spectrum
The differential diagnosis of uterine PEComa versus LMS is an area of diagnostic controversy. The diagnosis of uterine PEComa is particularly challenging because of the relative frequency of uterine smooth muscle tumours (SMTs). By middle age, up to 80% of women are believed to have uterine leiomyomas (LM).93 ,94 PEComas express a myoid immunophenotype and may have spindled morphology and therefore, it is unsurprising that they share many features with common SMTs such as LM and LMS.
While evidence of a myomelanocytic immunophenotype was initially considered a discriminatory feature in favour of PEComa, at least focal positivity for melanocytic markers such as HMB-45, Melan-A and MiTF has been demonstrated in uterine SMTs.95–97 In one study of five LMS with spindled and epithelioid morphology, the majority of HMB-45-positive areas exhibited clear cell morphology, but some HMB-45 positivity was also demonstrable in spindled areas.96 Evidence of the phenotypic plasticity of LMS is illustrated by a report of the development of a clear cell, diffusely HMB-45-positive, predominantly SMA-negative metastasis from a HMB-45-negative primary epithelioid LMS.95 Cathepsin K was recently promoted as a useful IHC marker for the differential diagnosis between PEComa and LMS, but further study has demonstrated cathepsin K expression in a proportion of LMS.98
Although the uterus is now the most commonly reported extrarenal site for PEComa,14 ,15 there has been some controversy as to whether uterine PEComa is a distinct entity.14 ,15 ,21 ,95–97 ,99 Evidence from four TSC patients with uterine PEComa, in whom the uterine tumour developed in the context of PEComatosis involving multiple gynaecological sites16–18 ,88 appears to confirm the existence of uterine PEComa. However, it is unclear whether a subset of tumours currently diagnosed as sporadic uterine PEComas actually represent uterine SMTs with variant histological and/or IHC features.
There are several reasons for caution when considering a diagnosis of uterine PEComa. First, there is overlap between the histological and IHC features of PEComas and uterine SMTs. While features such as epithelioid appearance, vascular architecture and the presence of spider cells, multinucleate giant cells and macronucleoli are proposed as being characteristic of PEComa,20 their use in distinguishing PEComa from uterine SMTs has not been validated. While classical spindled uterine LMS is a straightforward diagnosis in most cases, purely epithelioid LMS (rare) and, more commonly, mixed spindled and epithelioid LMS frequently pose diagnostic dilemmas due to their histopathological overlap with PEComas. As a result, there is variation between pathologists in the diagnosis of PEComa.
Second, criteria for malignancy in uterine LMS are well established, but much less so for PEComa. As discussed, the prognostic system for PEComa is in evolution as it is based on retrospective analyses of small tumour cohorts.20 ,53 A uterine tumour on the morphological spectrum between SMT and PEComa which demonstrates features such as infiltrative margin and increased mitotic activity may be labelled as benign or malignant, according to the classification of the tumour as a SMT or PEComa, respectively. This has major implications for the patient in terms of the risk of overtreatment, inappropriate treatment (eg, mTOR pathway inhibition) and psychological morbidity.
Third, there is little available molecular data. While several small studies of both TSC-associated and sporadic renal AML have confirmed the importance of TSC2 loss-of-function, it has been shown in only three uterine PEComas.39 ,40
Four patients with gynaecological PEComa have been reported to have received mTOR inhibitors (sirolimus and temsirolimus);69 ,78 ,100 follow-up is available on two patients, who died of their disease, at 9 and 10 months, respectively. In none of these four cases was LOH or mutation of TSC2 demonstrated nor was activation of the mTOR pathway confirmed. In our opinion, due to interpathologist variation and uncertainty in the diagnosis of uterine PEComa, all tumours accepted into clinical trials of mTOR pathway inhibitors should have molecular analysis to confirm loss of TSC2 and/or loss of function of tuberin with mTOR pathway activation.
Conclusion
What is the practicing pathologist to do when faced with a mixed spindled/epithelioid gynaecological tract tumour with mixed myoid and melanocytic marker expression? Fadare99 proposed that morphologically conventional SMTs be labelled as such, even if they focally express melanocytic markers (this should be noted in the report), while classical PEComa should also be diagnosed as such. He proposes that non-classical epithelioid mesenchymal tumours should be labelled as epithelioid tumours of UMP. In our opinion, it would be helpful to include a note in the pathology report explaining the basis for diagnostic uncertainty when using this term. For PEComas, the use of the modified-Folpe and Schoolmeester criteria may provide additional prognostic information to aid patient management. While these guidelines may help in the majority of cases, the ultimate question remains as to what constitutes a PEComa of the gynaecological tract outside of the setting of TSC, and how it can be reproducibly differentiated from SMTs. It is likely that molecular genetic studies will give us greater insight into the nature of gynaecological tract PEComa and its relationship to uterine SMTs and help resolve this ongoing diagnostic dilemma.
Take home messages
-
PEComas of the gynaecological tract are rare tumors, the majority of which are sporadic (not associated with tuberous sclerosis complex) uterine tumors with a mixed myo-melanocytic immunophenotype. Unusual tumor variants include sclerosing PEComa and TFE3-translocation associated PEComa.
-
The prognostic classification system for PEComa of the gynaecological tract is a work in progress. The modified-Folpe system described in this paper is a potentially useful tool for prognostic assessment.
-
The pathological differential diagnosis between uterine PEComa and leiomyosarcoma variants remains a challenge. Future molecular analysis of sporadic uterine PEComa to test the hypothesised role of TSC2 mutation in tumor development will likely provide valuable insights.
References
Footnotes
-
Handling editor Cheok Soon Lee
-
Contributors Preparation of initial draft, tables and figures: NC. Review and revision of manuscript and approval of final version of manuscript for submission: all authors.
-
Competing interests None.
-
Provenance and peer review Not commissioned; internally peer reviewed.