Article Text

Abstract
This article reviews the main molecular alterations involved in endometrial carcinoma. Five molecular features (microsatellite instability, and mutations in the PTEN, k-RAS, PIK3CA and β-catenin genes) are characteristic of endometrioid carcinomas, whereas non-endometrioid carcinomas show alterations of p53, loss of heterozygosity (LOH) on several chromosomes, as well as other molecular alterations (STK15, p16, E-cadherin and C-erb B2). The review also covers the phenomenon of apoptosis resistance, as well as the results obtained from cDNA array studies, and the perspectives for targeted therapies. A group of practical applications of molecular pathology techniques are also mentioned: diagnosis of hereditary non-polyposis colon cancer syndrome in patients with endometrial carcinoma; evaluation of precursor lesions; prognosis; diagnosis, particularly for synchronous endometrioid carcinomas of the uterus and the ovaries; and targeted therapies.
Statistics from Altmetric.com
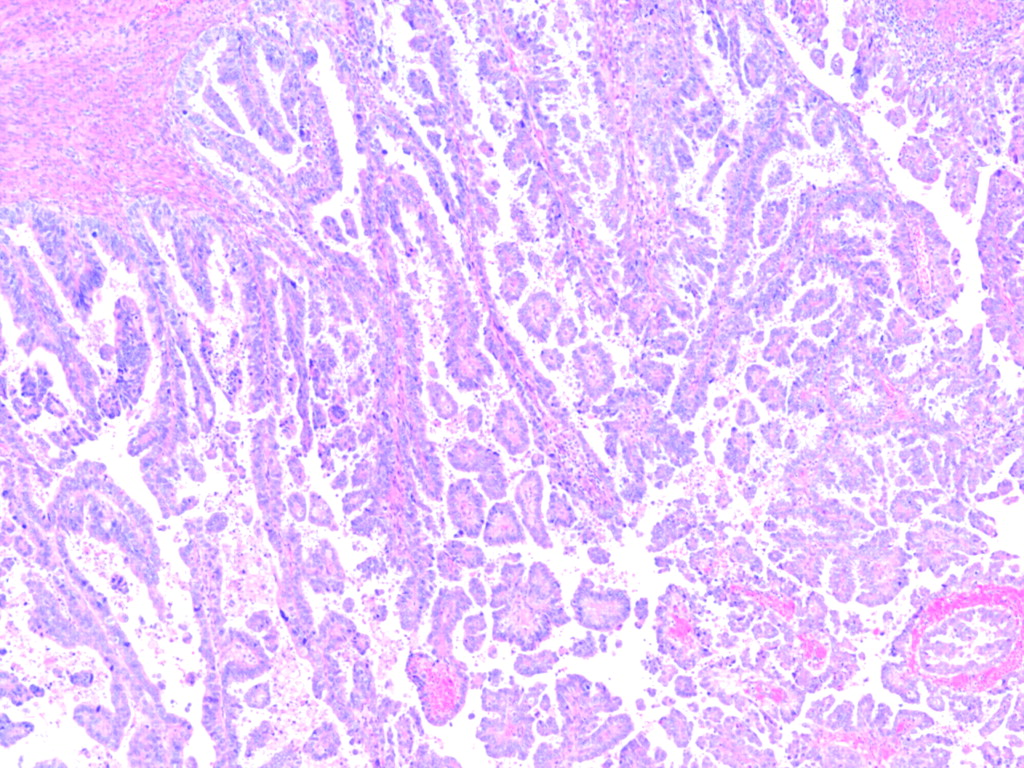
Bockman first described the two main clinicopathological variants of endometrial carcinoma (EC) (table 1).1 Type I tumours are low-grade and oestrogen-related endometrioid carcinomas (OEC) (fig 1) that usually develop in perimenopausal women and coexist or are preceded by complex and atypical endometrial hyperplasia. On the other hand, type II tumours are non-endometrioid (NOEC) carcinomas, very aggressive tumours, unrelated to oestrogen stimulation, arising occasionally in endometrial polyps or from precancerous lesions developing in atrophic endometrium, that mainly occur in older women. NOEC are usually serous (fig 2) or clear cell carcinomas. It has also been shown that the molecular alterations involved in the development of OEC carcinomas are different from those of NOEC carcinomas (table 2).2 3 While OEC show microsatellite instability (MI), and mutations in the PTEN, k-RAS, PIK3CA and β-catenin genes, NOEC exhibit alterations of p53, loss of heterozygosity (LOH) on several chromosomes, as well as other molecular alterations (STK15, p16, E-cadherin and C-erb B2).

Microscopic appearance of a type I, endometrioid carcinoma.

Low-power view of a non-endometrioid carcinoma (serous-type).
Clinical and pathological features of endometrial carcinoma, types I and II
Molecular features of endometrial carcinoma, types I and II
Molecular pathology of endometrioid carcinomas
Microsatellite instability (MI) is seen in cancers (colonic, endometrial and others) of patients with hereditary non-polyposis colon cancer (HNPCC), but is also seen in 25–30% of sporadic EC.4 5 6 7 8 HNPCC patients with EC have an inherited germ-line mutation in MLH-1, MSH-2, MSH-6 or PMS-2 (“first hit”); but EC develops only after the instauration of a deletion or mutation in the contralateral MLH-1,MSH-2, MSH-6 or PMS-2 allele (“second hit”) in endometrial cells. Once the two hits occur, the deficient mismatch repair role of the gene (MLH-1, MSH-2, MSH-6 or PMS-2) causes the acquisition of MI, and the development of the tumour. In sporadic EC, MLH-1 inactivation by promoter hypermethylation is the main cause of mismatch repair deficiency,9 which usually occurs at the precursor (atypical hyperplasia) lesion.10 Thus, MLH-1 hypermethylation is an early event in the pathogenesis of OEC, which precedes the development of MI. The prognostic significance of MI is under debate, but there is some convincing evidence suggesting association with favourable outcome.
The instauration of MI, the so-called mutator phenotype, leads to subsequent accumulation of myriads of mutations. Short-tandem repeats, like microsatellites, are particularly susceptible to mismatch repair alterations, but they are predominantly located in non-coding DNA sequences (fig 3); and the presence of subtle mutations (insertions or deletions) does not result in the production of abnormal proteins. However, some small short-tandem repeats, like mononucleotide repeats, are sometimes located within the coding sequence of some important genes (BAX, IGFIIR, hMSH3 and hMSH6). Mutations in these tracts are interpreted as secondary events in the mutator phenotype pathway in cancers with MI, probably responsible for tumour progression.11 12 13 14 15 16

Two examples of PIK3CA mutations in endometrial carcinomas (helicoidal and catalytic domains).
The tumour suppressor gene termed PTEN, located on chromosome 10q23.3, is frequently abnormal in endometrial carcinomas.17 18 19 20 21 22 23 24 25 LOH at chromosome 10q23 occurs in 40% of EC. Somatic PTEN mutations are also common in EC, and they are almost exclusively restricted to OEC, occurring in 37–61% of them. Interestingly, several groups have found a concordance between MI status and PTEN mutations; the mutations occur in 60–86% of MI positive OEC, but in only 24–35% of the MI negative tumours. PTEN mutations have been detected in endometrial hyperplasias with and without atypia (19% and 21%, respectively). Moreover, identical PTEN mutations have been detected in hyperplasias coexisting with MI positive OEC which suggests that PTEN mutations are early events in the development of OEC. Data are controversial regarding the prognostic significance of PTEN mutations in OEC, but there are some results that suggest association with favourable prognostic factors.
Mutations in PIK3CA, which codes for the p110α catalytic subunit of PI3K, have been described in various tumours and may contribute to the alteration of the PI3K/AKT signalling pathway in OEC.26 27 28 29 PI3K is a heterodimeric enzyme consisting of a catalytic subunit (p110) and a regulatory subunit (p85).The PIK3CA gene, located on chromosome 3q26.32, codes for the p110α catalytic subunit of PI3K. The mutations are predominantly located in the helical (exon 9) and kinase (exon 20) domains (fig 4). Oda et al described mutations in PIK3CA gene in endometrial carcinomas for the first time. In this series, PIK3CA mutations occurred in 36% of the cases, and coexisted frequently with PTEN mutations. Subsequent studies have shown that PIK3CA mutations are frequent in OEC, in association with invasion, and adverse prognostic factors such as blood vessel invasion.29

Microsatellite instability in an endometrioid carcinoma in a patient with hereditary non-polyposis colon cancer (HNPCC). Capillary electrophoresis shows deletion in mononucleotide tract BAT-26 in tumour DNA (T), in comparison with normal DNA (N).
The RAS-RAF-MEK-ERK signalling pathway plays an important role in tumourigenesis. The frequency of k-RAS mutations in OEC ranges between 10% and 30%.30 31 In some series, K-RAS mutations have been reported to be more frequent in OEC showing microsatellite instability. During tumourigenesis, activated RAS is usually associated with enhanced proliferation, transformation and cell survival. BRAF, another member of the RAS-RAF-MEK-ERK pathways, is very infrequently mutated in EC. RAS effectors like RASSF1A are supposed to have an inhibitory growth signal, which needs to be inactivated during tumourigenesis. Contradictory results between RASSF1A inactivation and K-RAS mutation have been obtained in different types of tumours. They were mutually exclusive events in colorectal and pancreatic cancer, but the correlation was not significant in lung cancer. Recent studies from our group have shown that RASSF1A inactivation by promoter hypermethylation may contribute significantly to increased activity of the RAS-RAF-MEK-ERK signalling pathway.32
The β-catenin gene (CTNNB1) maps to 3p21. β-Catenin appears to be important in the functional activities of both APC and E-cadherin. β-Catenin is a component of the E-cadherin-catenin unit, very important for cell differentiation, and maintenance of the normal tissue architecture. β-Catenin is also important in signal transduction. Increased cytoplasmic and nuclear levels of β-catenin produce transcriptional activation through the LEF/Tcf pathway. The APC protein down-regulates β-catenin levels by cooperating with the glycogen synthase kinase 3β (GSK-3β), inducing phosphorylation of the serine-threonine residues coded in exon 3 of the β-catenin gene (CTNNB 1), and its degradation through the ubiquitin-proteasome pathway. Mutations in exon 3 of β-catenin result in stabilisation of the protein, cytoplasmic and nuclear accumulation, and participation in signal transduction and transcriptional activation through the formation of complexes with DNA binding proteins. Mutations in exon 3 of CTNNB1 with nuclear accumulation of β-catenin occur in 14–44% of EC.33 34 35 36 37 38 They appear to be independent of the presence of MI, and the mutational status of PTEN and k-RAS. In all cases, the mutations were homogeneously distributed in different areas of the tumours, which suggests that they do play a role in early steps of endometrial tumourigenesis. In fact, alterations in β-catenin have been described in endometrial hyperplasias that contain squamous metaplasia (morules). Although there was a good correlation between CTNNB1 mutations and β-catenin nuclear immunostaining, the presence of a cytoplasmic and nuclear β-catenin immunoreactivity in some ECs that did not show a mutation in CTNNB suggests that alterations in other genes of the Wnt/β-catenin/LEF-1 pathway may be responsible for the stabilisation and putative transcription activator role of β-catenin in these tumours. Data are controversial regarding the prognostic significance of β-catenin mutations in EC, but they probably occur in tumours with good prognosis.
Molecular pathology of non-endometrioid carcinomas
In contrast to OEC, NOEC show p53 mutations (90%), inactivation of p16 (40%) and E-cadherin (80–90%), c-erbB2 amplification (30%), alterations in genes involved in the regulation of the mitotic spindle checkpoint (STK-15) and loss of heterozygosity at multiple loci, reflecting the presence of chromosomal instability.39 40 41 42 43 44 45 While p53 mutations occur in 90% of NOEC, they are only present in 10–20% of OEC, which are mostly grade 3 tumours. The p53 protein can induce apoptosis or prevent a cell from dividing if there is DNA damage. Mutation of the p53 gene diminishes the cell’s ability to repair damage to DNA before entry to S-phase, leading to a greater chance that mutations will be fixed in the genome and passed to successive generations of cells. Inactivation of the cell cycle regulator p16 is also more frequent in NOEC (40%) than in OEC (10%). The underlying mechanism is not clear, but probably involves deletion and promoter hypermethylation. Reduced expression of E-cadherin is frequent in EC, and may be caused by LOH or promoter hypermethylation. In fact, LOH at 16q22.1 is seen in almost 60% of NOEC, but in only 22% of OEC. C-erbB2 overexpression and amplification are also seen more frequently in NOEC (43% and 29%) than in OEC. However, the most typical molecular feature of NOEC is chromosomal instability. This phenomenon is characterised by the presence of widespread chromosomal gains and losses, which reflect the presence of aneuploidy. cDNA arrays have demonstrated that NOEC usually show up-regulation of genes (STK-15, BUB1, CCNB2) that are involved in the regulation of the mitotic spindle checkpoint. One of them, STK-15, which is essential for chromosome segregation and centrosome functions, is frequently amplified in NOEC.
None of the five main alterations of OEC (MI, and mutations in PTEN, k-RAS, PIK3CA and β-catenin) plays a significant role in NOEC. However, the occasional detection of these molecular alterations in NOEC shows that NOEC might develop by following two different pathways: (1) de novo, through p53 mutations, LOH at several loci, and some other, still unknown, gene alterations; or (2) dedifferentiation from pre-existing OEC. This hypothesis would explain the existence of mixed OEC–NOEC, and the presence of MI, as well as alterations in PTEN, K-RAS or β-catenin in NOEC. According to this point of view, de novo NOEC (by far the most common situation) would fulfil the clinicopathological and molecular features of NOEC (pure papillary serous or clear cell type morphology, old age, absence of oestrogen stimulation, lack of pre-existing endometrial hyperplasia, p53 mutations, lack of MI or PTEN mutations), whereas dedifferentiated NOEC would exhibit overlapping features with OEC (mixed NOEC–OEC morphology, early age of presentation, evidence of oestrogen stimulation or pre-existing hyperplasia, coexistence of p53 mutations, MI or PTEN mutations).
Apoptosis resistance in EC
Deregulation of apoptosis plays an important role in development and progression of cancer. Moreover, cells resistant to apoptosis are likely to escape the immune surveillance, but they may be also resistant to therapy. Apoptosis can be initiated by two main mechanisms: the intrinsic pathway, which has its origin in the mitochondria, and the extrinsic apoptotic pathway, triggered by the activation of death receptors situated in the cell surface.
The “extrinsic pathway” is activated by ligand-bound death receptors such as tumour necrosis factor (TNF), Fas or TRAIL receptors. After ligand binding, the activated death receptors recruit an adaptor protein named Fas associated death domain (FADD). FADD consists of two protein interaction domains: a death domain (DD) and a death effector domain (DED). FADD binds to the receptor through interactions between DDs and to pro-caspase-8 through DED interactions to form a complex at the receptor called the death inducing signalling complex (DISC). Recruitment of caspase-8 through FADD leads to its auto-cleavage and activation. Active caspase-8 in turn activates effector caspases such as caspase-3, causing the cell to undergo apoptosis by digesting upwards of a hundred or so proteins. One of the key regulators of this signalling is c-FLIP, which shares a high degree of homology with caspase-8 but lacks protease activity. In some type of cells this relatively simple pathway is enough to trigger apoptosis, but in other types of cells, the death receptor apoptotic pathway requires mitochondrial amplification. The BH3-only protein Bid is cleaved by caspase-8 and is then translocated to the mitochondria to activate the intrinsic pathway, thus connecting the two caspase activation pathways and amplifying the death receptor apoptotic signal. Thus, alterations in the mitochondrial (“intrinsic”) pathway may affect the ability to induce apoptosis by death receptors.
There is much evidence suggesting that alteration of apoptosis is important in development and progression of EC. Several of the molecular abnormalities that have been detected in EC may be associated with apoptosis deregulation. As mentioned before, activation of the PI3K/AKT pathway suppresses apoptosis triggered by various stimuli. Moreover, the recent evidence that NF-kB activation is frequent in endometrial carcinoma may explain the presence of apoptosis resistance by activation of target genes such as FLIP and Bcl-XL.46 p53 alterations, which are characteristic of NOEC, may also occur in endometrioid tumours, particularly in those neoplasms showing overlapping features between types I and II tumours; and they may have an impact in apoptosis at several different levels. Also, members of the Bcl-2 family of genes are abnormal in endometrial carcinoma. For example, BAX is a target gene for mutations in OEC with microsatellite instability, and may have a role in resistance to apoptosis in these tumours. Finally, several other proteins involved in apoptotic control (survivin) have also been shown to be abnormal in endometrial carcinoma.47 An important protein responsible for apoptosis resistance in endometrial carcinoma is FLIP. FLIP expression is frequent in endometrial carcinomas.48 A direct evidence of the role of FLIP in TRAIL apoptosis resistance on endometrial carcinoma cells is provided by treatment with specific siRNA targeting FLIP (fig 5). Transfection of endometrial carcinoma cell lines with FLIP siRNA results in a marked decrease in cell viability, after TRAIL exposition. This is accompanied by activation of both caspase-8 and caspase-3, suggesting activation of the extrinsic pathway. Recent studies have shown that FLIP may be regulated by a cellular complex, CK2.49
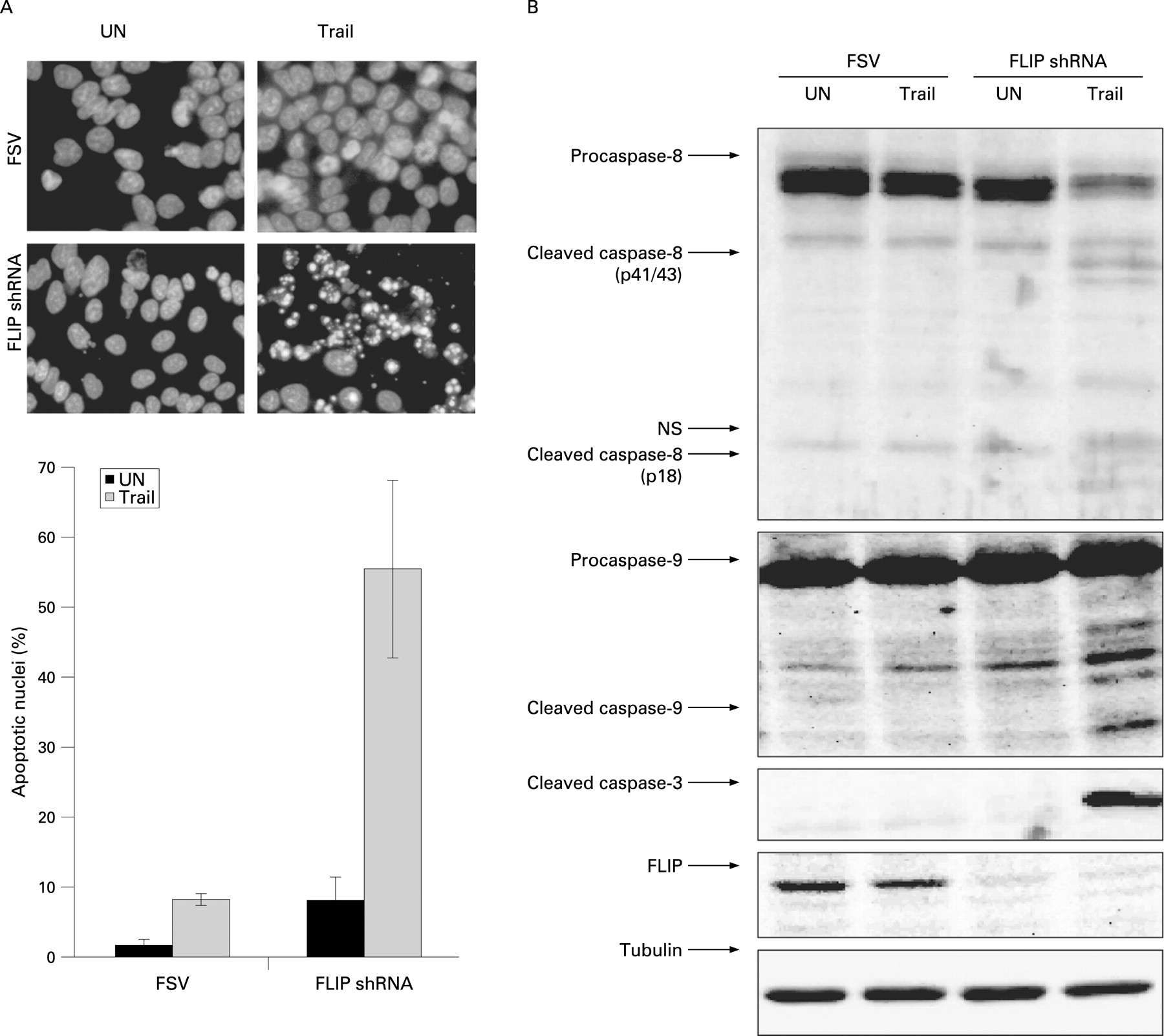
Knock-down of endogenous FLIP protein levels sensitises Ishikawa (IK) endometrial carcinoma cells to TRAIL-induced apoptosis. (A) IK cells were transduced with lentiviruses carrying short hairpin RNA against cellular FLIP or with an empty vector (FSV). After three days post-infection cells were cultured in the presence of TRAIL (50 ng/ml for 12 hours). Top, nuclei displaying apoptotic morphology were visualised by Hoechst staining (representative panel). Bottom, quantification of apoptotic nuclei. (B) Delivery of TRAIL into culture media triggers activation of caspases-8, -9 and -3 in FLIP-silenced IK cells. Three days post-infected-IK cells were subjected to TRAIL stimuli or left untreated (UN). After 12 hours cells were lysed and protein extracts were analysed by western blot with antibodies to the indicated caspases. Membranes were reprobed with tubulin to ensure equal protein amounts. NS, non-specific bands.
cDNA array studies
cDNA array analysis has demonstrated that the expression profiling of OEC is different from that of NOEC.50 51 52 53 54 55 56 57 58 59 60 In one study, 191 genes exhibited a >2-fold differences between 10 OECs and 16 NOECs. One of the genes, TFF3, was significantly up-regulated in OECs, while increased expression of FOLR was seen in NOECs. In another study, a different expression profile was seen between OEC and NOEC; the differences involved 66 genes. Interestingly, oestrogen-regulated genes were up-regulated in OEC, whereas NOEC showed increased expression of genes involved in the regulation of the mitotic spindle checkpoint. A third study demonstrated differential expression of 1055 genes between OECs and serous carcinomas. Genes up-regulated in serous carcinomas were IGF2, PTGS1 and p16, while genes up-regulated in OEC included TFF3, FOXA2 and MSX2. Another study identified 315 genes that statistically differentiated OEC from NOEC. Moreover, a different expression profile was also found between EC associated with microsatellite instability and stable EC.54 Interestingly, two members of the secreted frizzled related protein family (SFRP1 and SFRP4) were more frequently down-regulated in EC with microsatellite instability. In another study, it was seen that the tumours (ovarian and uterine) with β-catenin alterations, showed common gene expression profile. A group of investigators have also identified, by c-DNA array studies, up-regulation of RUNX1/AML1 and ERM/ETV5 in EC, and suggested an implication of such genes in myometrial invasion.56 One study compared the expression profiles of similar histological subtypes of ovarian and endometrial carcinomas, and showed that clear cell carcinomas had a very similar profile, regardless of the organ of origin. In contrast, differences were seen when comparing endometrioid and serous carcinomas of ovarian and endometrial origin.57
Targeted therapies in endometrial carcinoma
There are several different signalling pathways that are good candidates for targeted therapy in EC.58 As mentioned before, the PI3K/AKT pathway is the most frequent abnormal signalling pathway in OEC, often resulting from mutations in the tumour suppressor gene PTEN, and activating mutations in PIK3CA. The importance of the PI3K/AKT survival pathway in EC raises the possibility that PI3K inhibitors, such as wortmannin and derivatives, may be used as potential anticancer agents. In fact, a decrease of AKT phosphorylation and increased apoptosis are seen in mutated PTEN human endometrial cancer cells in the presence of PI 3-K inhibitor. Of particular interest among AKT targets is the downstream effector mTOR. The TOR family of proteins has pleiotropic functions, and participates in the regulation of the initiation of mRNA transcription and protein translation in response to intracellular concentrations of amino acids and other essential nutrients, in the organisation of the actin cytoskeleton, membrane trafficking, protein degradation, PKC signalling and ribosome biogenesis. mTOR regulates essential transduction pathways and is involved in coupling growth stimuli to cell cycle progression. AKT activates mTOR via direct phosphorylation of TCS2 and by the inhibition of AMPPK, thereby activating Rheb and mTOR-Raptor activity. On activation, mTOR-Raptor activates S6K and inhibits 4EBP1 to accelerate mRNA translation. However, the mTOR pathway can also be activated by other mechanisms including activation of tyrosine kinase receptors (epidermal growth factor receptors EGFR1-4, PDGFR, KIT, IGFR) and Ras. Moreover, loss of function of p53 may also result in activation of mTOR. mTOR inhibitors (rapamycin and rapamycin derivatives) have been recently developed as potential anticancer agents. Tumours associated with PTEN inactivation, like EC, are particularly susceptible to the therapeutic effects of mTOR inhibitors. Several mTOR inhibitors are available for clinical trials: the prototype rapamycin and three rapamycin derivatives, CCI-779 (temsirolimus), RAD001 (everolimus) and AP23573. Both CCI-779 (temsirolimus), and RAD001 (everolimus) are currently being tested in two phase II trials, in recurrent endometrial cancer.58 PTEN+/− mice is a good model to test sensitivity of EC to anticancer drugs, because they develop complex atypical hyperplasia and endometrial carcinoma. Pharmacological inhibition of mTOR by CCI-779 in PTEN+/− mice has shown reduced neoplastic proliferation, tumour size and S6K activity.59
Tyrosine kinase receptors are also good targets for anticancer therapies. The epidermal growth factor family and its growth factors are known to play critical roles in cell growth and differentiation. The epidermal growth factor family is comprised of EGFR (ErbB1), HER-2/neu (ErbB2), HER-3 (ErbB3) and HER-4 (ErbB4). EGFR and HER-2/neu have been shown to be highly expressed in normal endometrium and overexpressed in endometrial cancer. Increased expression of EGF related protein and EGFR may contribute to a drug resistant phenotype. EGFR has intrinsic tyrosine kinase activity which is activated on ligand binding. Downstream serine/threonine kinase cascades are then induced, leading to cell proliferation and the inhibition of apoptosis. Tyrosine kinase inhibitors prevent the phosphorylation of MAP kinase Erk1/2, MAP kinase MEK 1/2, CDK 1, AKT, RAF 1 and Rb-1. Inhibition of EGFR with monoclonal antibodies leads to growth arrest, and a similar and potentially synergistic effect is anticipated with inhibition of EGFR tyrosine kinase activity. Potential agents are iressa (ZD1839), Herceptin (trastuzumab), and lapatinib (GW572016). The signalling pathways of EGFR, under the effect of EGF and ZD1839, were evaluated in EC cell lines; results showed that both OEC and NOEC have the capacity to respond to EGFR inhibition, but the response of NOEC may be limited by the constitutive activation of other signalling pathways. ZD1839 has been studied as a single agent in a phase II trial (GOG 229C) of women with advanced EC. Preliminary data showed one complete response and stable disease in several patients after 6 months.
The EGFR pathway may be induced by steroid hormones, which is the principal growth promoting mechanism in the endometrium. Oestrogen and progesterone are the most important steroid hormones that modulate endometrial cell proliferation and differentiation, respectively, and their receptors are expressed in about 80% of OEC, although the proportion may decrease with a loss of differentiation. NOEC are not responsive to hormones and may not express oestrogen and progesterone receptors. Medroxyprogestererone acetate has been studied in patients with EC. Two large GOG trials evaluating oral progestins in these patients showed an overall response rate of 15–25%, with median progression-free survival of less than 4 months and overall survival of less than 11 months.
As discussed before, apoptosis is a key process in the regulation of cellular homoeostasis, which may be subjected to therapeutic targeting. Proteasome inhibitors are currently used as chemotherapeutic drugs because of their ability to trigger cell growth arrest or apoptosis on several tumours. In many different types of tumour cells, bortezomib and other proteasome inhibitors cause cell death by blocking NF-kB activity. However, in EC, proteasome inhibitors induce cell death, but, instead of blocking NF-kB, they increase its transcriptional activity. Proteasome inhibitors induce phosphorylation of IKKα/β, phosphorylation and degradation of IkBα and phosphorylation of p65 NF-kB subunit on serine 536.60 61 Proteasome-inhibitor induced cell death was accompanied by activation of caspases and apoptotic nuclear morphology.
Sorafenib (BAY 43-9006, Nexavar) is a potent, orally administered receptor tyrosine kinase inhibitor with antiproliferative and antiangiogenic activities. Sorafenib was originally described as an inhibitor of B- and c-RAF kinase, but also has activity against several receptor tyrosine kinases, including vascular endothelial growth factor receptor 2 (VEGFR2), platelet-derived growth factor receptor (PDGFR), FLT3, Ret and c-Kit. The antitumour activity of sorafenib in hereditary colon cancer may be attributed to inhibition of tumour angiogenesis (VEGFR and PDGFR) and direct effects on tumour cell proliferation/survival (Raf kinase signalling-dependent and signalling-independent mechanisms). A recent multicentre phase II trial in patients with advanced/recurrent EC has shown that sorafenib may result in clinical benefit for a minority of EC patients. Preliminary results from our group have shown that pharmacological inhibition of B-RAF by sorafenib sensitised EC cells to TRAIL-induced apoptosis, by down-regulating FLIP.
Histone acetylation is one of the mechanisms involved in the epigenetic control of gene expression. Altered histone acetylation in cancer cells may be responsible for abnormal expression of oncogenes and tumour suppressor genes. Histone deacetylase inhibitors (HDACI) are promising anticancer drugs. In cancer cells, HDACI cause cell-cycle arrest, apoptosis and differentiation. HDACI cause de-repression of genes whose reactivation would promote an anti-proliferative effect. Examples of genes up-regulated by HDACI are p21, TRAIL-R2, p19ARF, Bmf and Rap1. Paradoxically, HDACI cause down-regulation of important genes such as thymidylate synthetase, Bcr-Abl and c-Myc. Interestingly, microarray studies have shown that HDACI treatment leads to changes in RNA levels of a surprisingly small number of genes (around 2–5% of the genome). Some studies have suggested that HDACI may be more effective in tumours expressing mutant p53. Preclinical studies using HDACI in endometrial carcinoma cell lines have provided interesting results.62 63 For example, HDACI have been shown to induce differentiation of endometrial carcinoma cell lines, which resemble normal endometrial epithelial cells in the absence of ovarian steroid hormones. Moreover, some studies have shown that HDACI had an important growth inhibitory effect on EC cell lines, by decreasing the proportion of cells in S phase, and increasing the proportion of cells in the G0-G1 and or G2-M phases of the cell cycle. There was also up-regulation of p21, p27 and E-cadherin, and down-regulation of Bcl-2 and cyclin-D1 and -D2. The growth-suppressor effects seem to be irrespective of the p53 gene status.
Practical aspects of the molecular pathology of endometrial carcinomas
Evaluation of the molecular features of EC is still an area of predominant interest in the field of research. However, there are some aspects that could have practical usefulness at present or in the near future; these are discussed below.
Diagnosis of HNPCC in a patient with EC
It is now accepted that immunostaining for mismatch repair genes (MLH-1, MSH-2, MSH-6 or PMS-2), as well as evaluation of microsatellite instability, is of obvious interest in studying EC patients with suspected features of HNPCC (young age, premenopausal status, coexistence with colonic or other HNPCC-associated tumours, strong family history). Results should be confirmed by identifying the germ-line mutation in the responsible gene (MLH-1, MSH-2, MSH-6 or PMS-2) in DNA obtained from normal tissue or peripheral blood.
Evaluation of precursor lesions
Some investigators have suggested an important role of PTEN in evaluation of EC precursor lesions. However, the reliability of PTEN immunostaining is still under debate, since problems of reproducibility have not been solved.64 The authors of this review have used MLH-1 immunostaining in evaluation of atypical complex hyperplasia in endometrial biopsies. As mentioned before, abnormal methylation of MLH-1 may be detected in atypical hyperplasias, which frequently coexist with adenocarcinoma in the hysterectomy specimen. For that reason, identification of negative staining for MLH-1 in a young, premenopausal patient with a diagnosis of atypical complex hyperplasia in a previous endometrial biopsy could be taken as an indirect evidence for risk of coexistent EC.
Prognosis
There is evidence suggesting that the presence of microsatellite instability, or mutations of PTEN and CTNNB-1 (β-catenin) could indicate a good prognosis for EC, while the presence of p53 abnormalities or PIK3CA mutations could suggest aggressive behaviour. However, accumulated data regarding the prognostic significance of these alterations are still scarce.
Diagnosis
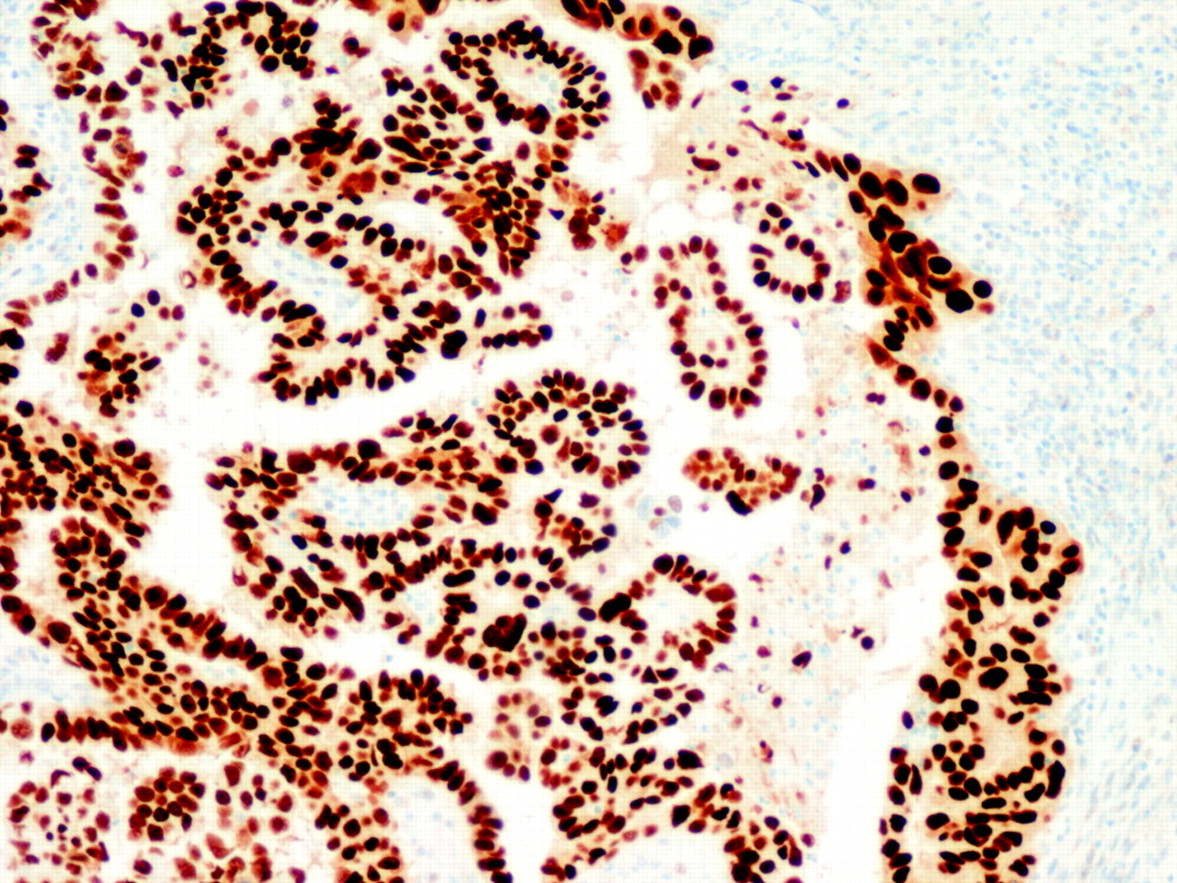
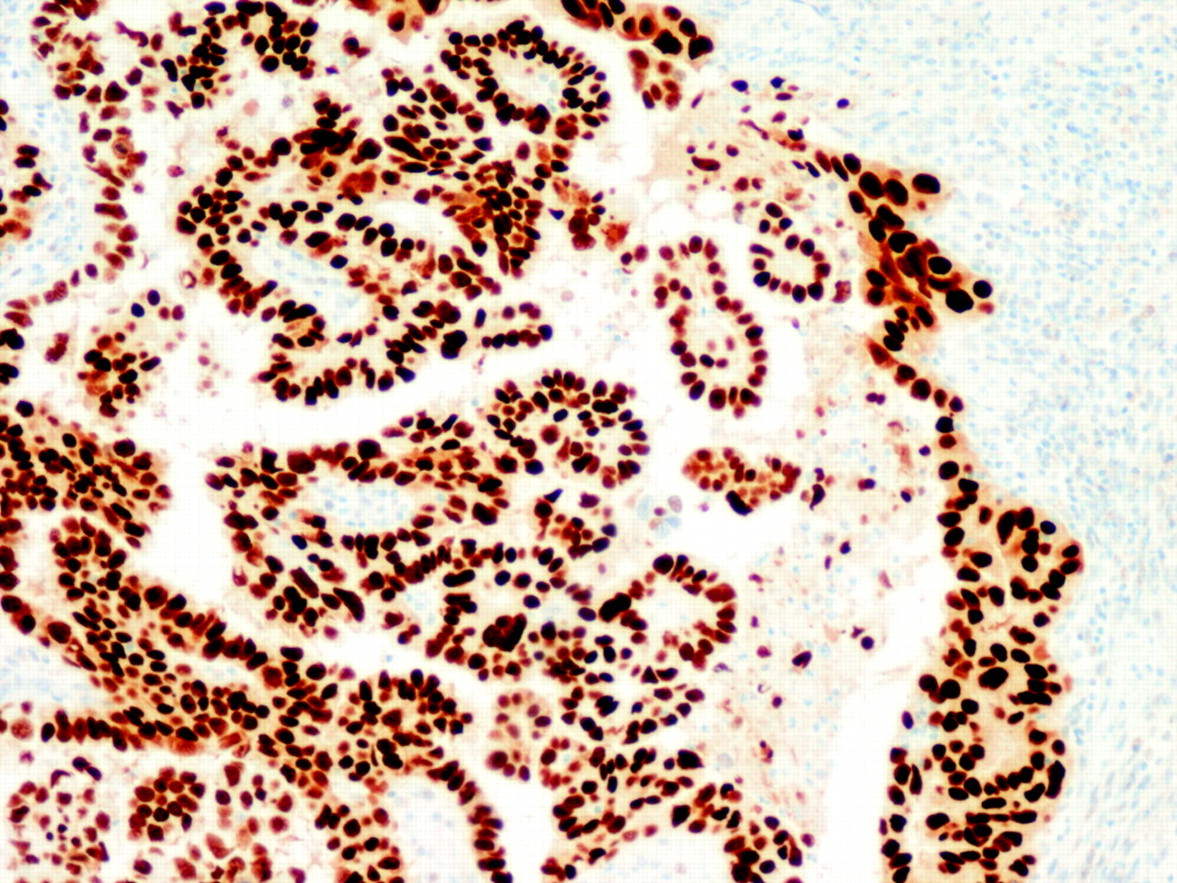
The diagnostic utility of the molecular alterations is still under debate. There are five alterations that are typical of OEC (microsatellite instability, and mutations in the PTEN, k-RAS, PIK3CA and β-catenin genes) and some others that are characteristic of NOEC (alterations of p53, LOH on several chromosomes, as well as alterations in STK15, p16, E-cadherin and C-erb B2), but there is some degree of overlapping. For that reason, their diagnostic usefulness is limited. Probably, p53 is the alteration that has more practical relevance, since NOEC shows frequent p53 expression (fig 6). It is important, however, to keep in mind that p53 may be altered in OEC, particularly in high grade tumours.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Strong p53 immunostaining in a non-endometrioid carcinoma (serous type).
One potential indication for assessment of molecular alterations in EC is the appropriate interpretation of simultaneous presence of endometrioid carcinoma in the endometrium and the ovaries. In the vast majority of these cases, there is coexistence of two independent primary tumours; but in some others the ovarian tumour is a metastasis from the endometrial carcinoma. It is very important to make the correct diagnosis, because the prognosis is related to the independent versus metastatic nature of the ovarian tumour. The differential diagnosis of these two possibilities relies on clinical and pathological features (bilaterality, pattern of growth, tubal invasion and coexistence with ovarian endometriosis). However, the identification of identical molecular features in both tumour sites strongly suggests that one is metastatic from the other.65
Targeted therapies
Although the indication of targeted therapies in EC is still under evaluation, it is possible that in the near future, we will be required to check molecular features of EC (particularly for widely disseminated and recurrent tumours) to suggest the most appropriate approach to treat the patient.
Take-home messages
There are two clinicopathological variants of endometrial carcinomas (endometrioid and non-endometrioid) that show specific molecular features and different gene expression profiles.
The main molecular features of endometrioid carcinomas are: microsatellite instability, and mutations in PTEN, k-RAS, PIK3CA and β-catenin genes. Although the clinical and prognostic relevance of each of these alterations has not been fully elucidated, there is evidence showing that microsatellite instability, and mutations in the PTEN and β-catenin genes would be associated with favourable outcome.
The main features of non-endometrioid carcinomas are p53 mutations, inactivation of p16 and E-cadherin, c-erbB2 amplification, alterations in genes involved in the regulation of the mitotic spindle checkpoint (STK-15) and loss of heterozygosity at multiple loci, reflecting the presence of chromosomal instability.
Targeting PI3K/AKT/mTOR, as well as genes involved in apoptosis resistance (proteasome inhibitors, B-RAF), and histone acetylation may play a role in the future treatment of patients with metastatic endometrial carcinoma.
REFERENCES
Footnotes
Funding Supported by grants FISPI060577, FISPI070304, FISPI070276, SAF2002-10529-E, Marató de TV3 2005-47, 2004XT00090 and RD06/0020/1034. XD holds a postdoctoral fellowship from the Fondo de Investigaciones Sanitarias, Ministerio de Sanidad y Consumo (CP05/00028). DL holds a predoctoral fellowship from the Fondo de Investigaciones Sanitarias, Ministerio de Sanidad y Consumo (FI05/00191). NE holds a predoctoral fellowship from the Fondo de Investigaciones Sanitarias, Ministerio de Sanidad y Consumo (FI08/00012) and Fundació Alicia Cuello de Merigó.
Competing interests None.
Ethics approval Ethics approval was obtained.