Article Text

Abstract
In the International Society of Paediatric Oncology renal tumour trials, preoperative chemotherapy has been successfully applied with resulting reduction of tumour rupture and increased favourable stage distribution of nephroblastoma. Postoperative treatment includes chemotherapy and sometimes radiotherapy in a risk-adapted approach based on histological sub-classification and stage of the tumour. However, preoperative chemotherapy alters the tumour's histological features and distribution of subtypes, and makes staging more difficult. The paper highlights the most common practical diagnostic difficulties that a pathologist is faced with in dealing with pretreated nephroblastomas. It emphasises the importance of a systematic, step-by-step analysis based on adequately sampled material, in order to accurately sub-classify a nephroblastoma as a low, intermediate or high risk tumour and assign its genuine stage. Finally, it outlines the standard operating procedure for submission of renal tumours for rapid central pathology review which allows the treating oncologists to apply the optimal treatment protocol.
Statistics from Altmetric.com
Renal tumours comprise 7–8% of all tumours in the first 15 years of life. Wilms' tumour (WT) or nephroblastoma is by far the most common (∼85% of cases), followed by renal cell carcinomas (∼3–5%), mesoblastic nephroma (∼3%), clear cell sarcoma of the kidney (∼3–4%), rhabdoid tumour of the kidney (∼2%) and miscellaneous rare tumours (∼2%).1 Accurate histological diagnosis and staging of these tumours are critical because their treatment and prognosis are very different. Since they are rare, they still represent a diagnostic and therapeutic challenge,2 and it is important to treat and study them through large national and international multicentre collaborative trials which include centralised pathological review in order to verify the diagnosis and stage of cases entered on the trials.
The first multicentre trial started in 1969 in the United States through the National Wilms' Tumour Study group (NWTS; now part of the Children's Oncology Group, COG) with the most important objective being the establishment of optimal treatment for WT. NWTS/COG have been treating WTs with surgery first, followed by, if necessary, postoperative chemotherapy and radiotherapy.3 On the other hand, preoperative therapy has been an essential part of the International Society of Paediatric Oncology (SIOP) treatment strategy since its first trials. In the first two trials (SIOP 1, 1971–74; and SIOP 2, 1974–76) preoperative radiotherapy was used. Since the third trial (SIOP 5, 1977–79) it has been replaced with preoperative chemotherapy which has shown very similar results in terms of preventing tumour rupture(s) and inducing more favourable stage distribution, with more stage I tumours requiring less postoperative therapy (SIOP 5, 1977–79; SIOP 6, 1980–86; SIOP 9, 1987–93; and SIOP 93 01, 1993–2001).4 Due to the different therapeutic approaches between the two groups, there are further differences in histological sub-classification and staging (see below), making a direct comparison of their results complicated.
In 1978, Beckwith and Palmer introduced a histological classification of primarily operated WTs with two main groups—anaplastic and non-anaplastic—which became a basis for their treatment in NWTS/COG trials.5 By studying the correlation between the histological features and survival of WTs that received preoperative chemotherapy from the early SIOP trials, it became apparent that they could be sub-classified into three treatment groups: favourable, standard and unfavourable histology groups.6 In the more recent classifications (SIOP 93 01 and the current SIOP 2001), they have been renamed into low risk, intermediate risk and high risk tumours (table 1).7 8 This concept of stratification of tumours into low, intermediate and high risk groups has later been followed in other tumours of childhood, including rhabdomysarcomas, neuroblastomas, hepatoblastomas and germ cell tumours.
The revised International Society of Paediatric Oncology (SIOP) working classification of renal tumours of childhood (2001)
Further analyses of WT subtypes from SIOP trials have resulted in removal from or addition of certain subtypes to the risk groups. So, for example, WT with fibroadenoma-like structures, which was earlier regarded as a favourable histology tumour, has disappeared from the new classification since it has been recognised that it represented a pattern of growth of some WTs rather than a distinct histological type.9 On the other hand, it has been shown that a tumour's response to preoperative chemotherapy is an indicator of good prognosis,10 so completely necrotic WT has been placed in the low risk tumour group (table 1).8 In the current SIOP 2001 trial (2001–), other WT types including epithelial, stromal and regressive (sub)type with >90% necrosis are being investigated since preliminary results have indicated that their prognosis is better than for other WT types from the intermediate risk group.11 12 In the same way, the presence of a certain amount of blastema after preoperative chemotherapy clearly indicates its non-responsiveness to chemotherapy, and blastemal type WT has been shown to be associated with poorer outcome13 and is therefore moved into high risk tumour group.8
According to the SIOP WT 2001 trial protocol, renal tumours in children are treated with preoperative chemotherapy consisting of two drugs given over a period of 4 weeks. Unlike in the rest of the SIOP, in the UK a histological diagnosis made on percutaneous cutting needle (“tru-cut”) biopsy is required before preoperative chemotherapy.14 15 Chemotherapy is followed by surgery and further chemotherapy and/or radiotherapy, if necessary, depending on the tumour's histological subtype and stage.
The pathologist has a critical role in the following:
Making an accurate histological diagnosis.
Assigning the tumour's histological subtype and risk group.
Making a precise evaluation of the abdominal stage of the tumour (even in children with stage IV disease, local staging is crucial in determining the use of radiotherapy).
For pathologists, there is one major disadvantage of preoperative chemotherapy—it significantly alters the histological features of WT, resulting in different histological patterns and distribution of subtypes from those treated with immediate surgery. In the earlier SIOP study, in the immediately operated WTs the most common subtype was mixed (45.1%), followed by blastemal (39.4%) and epithelial predominant (15.5%), whereas in tumours that received preoperative chemotherapy, the most common type was regressive (37.6%), followed by mixed (29.4%), stromal (14%), blastemal (9.3%) and epithelial predominant (3.1%); 6.6% of tumours were completely necrotic.13 Preoperative chemotherapy is more likely to destroy blastema and (less differentiated) epithelial elements, while it induces maturation especially in the stromal component, where rhabdomyoblastic differentiation is much more common than in primarily operated tumours. The typical chemotherapy-induced changes of treated WTs are a mixture of coagulative-type necrosis of small round cells or neoplastic tubules consisting of pink, necrotic nuclei, consistent with coagulative necrosis of blastemal cells or neoplastic tubules, fibrosis, hypocellular stroma containing foamy and/or haemosiderin-laden macrophages, and haemorrhage.
In order to subtype a WT, a pathologist has to evaluate it in a particular order, as follows:
Assess the percentage of necrosis/regressive changes caused by chemotherapy.
If only necrosis/regressive changes are present, with no viable tumour identified in an adequately sampled tumour (see below), it is regarded as completely necrotic WT.
If regressive changes comprise more than two-thirds of a tumour mass, it is a regressive type.
If regressive changes comprise less than two-thirds of a tumour mass, the viable tumour is sub-classified on the basis of the viable components present (blastemal, epithelial, stromal or mixed type).
If focal anaplasia is found, the tumour should still be sub-classified on the basis of other components.
The criteria for subtyping a nephroblastoma in the SIOP WT 2001 classification are detailed elsewhere.8 They include new definitions of certain WT types which have been introduced in order to re-emphasise that they differ from WTs that are treated with no preoperative chemotherapy (table 2). Therefore, it is very important not to confuse what is called epithelial or stromal type WT in SIOP WT 2001 classification with epithelial or stromal predominant WT in the NWTS/COG classifications. The SIOP WT 2001 criteria for epithelial or stromal type WT are that the epithelial or stromal component comprises more than 66% of the viable tumour, but the rest of the tumour may contain only up to 10% of blastema (if it contains more, than it is regarded as mixed type). On the other hand, in the NWTS/COG classification, the tumour is called epithelial or stromal predominant if the epithelial or stromal component comprises more than 66% of the tumour, irrespective of the remaining tumour component(s). So, for example, a WT consisting of 70% epithelial and 30% blastemal components would be regarded as epithelial predominant WT in NWTS/COG classification, whereas the same tumour would be not be regarded as epithelial but mixed type in the SIOP classification.
Histological criteria for Wilms' tumour subtyping in SIOP WT 2001 trial
Common problems in assessment of pretreated Wilms' tumours
In assessing a pretreated WT, a diagnostic pathologist is faced with a number of problems. Some which may significantly influence tumour's sub-classification and, consequently, treatment are as follows:
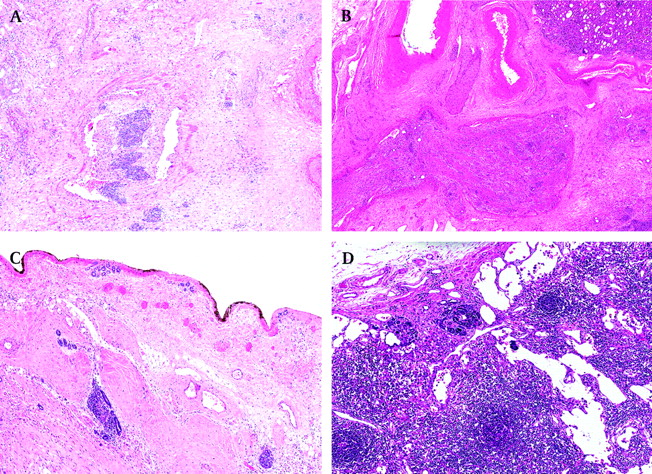
Completely necrotic tumour—was it a WT? Even in completely necrotic WTs, one can often see the contours of pre-existing tumour components (figure 1A,B). If not, look for nephrogenic rests which are only associated with WTs. Finally, if none of these is seen/found, it is still reasonable to assume that the tumour was a WT since other renal tumours usually do not respond so well to relatively mild preoperative chemotherapy that is given for WTs.
Occasional tubules and small foci of stroma in otherwise completely necrotic tumour (figure 1C). These findings are regarded as nephrogenic remnants and should not be taken into account for subtyping purposes, so the tumour should be classified as a completely necrotic type (low risk) rather than a regressive type (intermediate risk).
Necrotic versus viable tumour. There are no clear-cut criteria on the basis of which one could say with absolute certainty when the tumour is not viable any longer. It is relatively easy to recognise genuinely “dead” tumour where either “ghost” tumour cells (figure 1A, B) or foamy macrophages are usually seen, but it is not so easy when tumour cells are “dying” and their nuclear morphology is disappearing but is still vaguely recognisable (figure 1D). We regard such areas as non-viable and would not take them into account for subtyping a tumour.
Hypocellular tumour stroma or chemotherapy-induced changes. It is not always possible to distinguish genuine, hypocellular tumour stroma (figure 1E) from chemotherapy-induced changes, and one should look for other clues such as a finding of rare rhabdomyoblasts (figure 1F) (which would indicate that it is a genuine viable tumour stroma) or foamy macrophages (which would indicate that such areas represent chemotherapy-induced changes).
More or less than 66% blastema? (ie, blastemal type (high risk) versus mixed type (intermediate risk) with a lot of blastema). This is one of the most challenging dilemmas because of very serious implications for postoperative treatment and, again, there is no simple answer. The estimation of the percentage of tumour components is semi-quantitative, and by looking at the series of slides from a tumour, a pathologist builds a picture in his/her brain (like a jigsaw) and ends up with an estimate. The histological features of a WT may vary considerably, so sometimes it is necessary to estimate tumour components slide by slide, and add them up. But all these methods are subjective and sometimes one just has to rely on expert opinion (which is, admittedly, equally subjective).
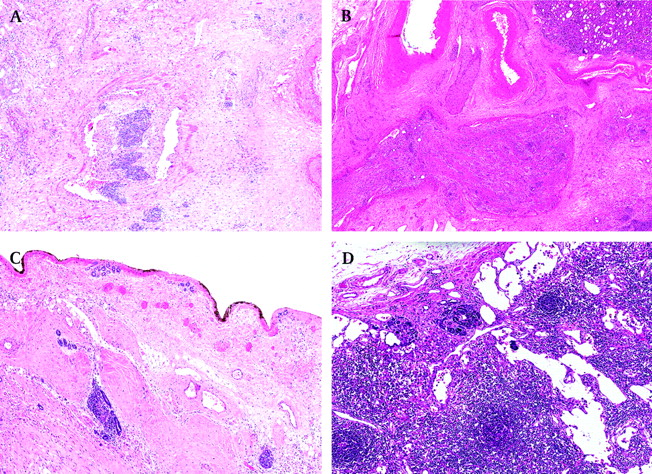
Anaplasia or not? To make a diagnosis of anaplasia, all three criteria have to be confirmed, including the presence of atypical tri/multipolar mitotic figures, marked nuclear enlargement, with diameters at least three times those of adjacent cells of the same type, and the presence of hyperchromatic tumour cell nuclei (figure 2A). If there are large hyperchromatic nuclei but no atypical mitoses, one should assess the overall mitotic activity. If it is low, it may be very difficult to find atypical mitoses and in such cases the finding of just one atypical mitosis is enough. Curiously, anaplasia still represents the biggest problem in Wilms' tumour histology (in the NWTS 5 series of anaplastic WTs, 39% cases were misdiagnosed by the institutional pathologists).16 Anaplasia may occur in the epithelial, blastemal or even stromal component of WT and it can be either focal or diffuse.17 The definition of focal anaplasia underlines the distribution of anaplasia which has been defined as the presence of a clearly defined focus within a primary intra-renal tumour, without evidence of anaplasia or prominent nuclear atypia in other sites (figure 2B). Very rarely, focal anaplasia may have more than one focus, but each of them has to be surrounded on all slides by non-anaplastic tumour. The finding of anaplasia in any other circumstances is regarded as diffuse anaplasia. Preoperative chemotherapy does not eradicate or generate anaplasia (although there are cases of bilateral non-anaplastic WTs treated with prolonged preoperative chemotherapy which eventually became anaplastic; personal experience). There are a number of histological findings which may mimic anaplasia such as fused or smudged masses of nuclear chromatin due to technical artefact, overlapping cells in thick sections, stain precipitate and circulating megakaryocytes. The most frequently observed pseudo-anaplastic changes are seen in the stromal rhabdomyoblasts which may exhibit large, hyperchromatic, multinucleated, bizarre nuclei as a response to chemotherapy (figure 2C,D).
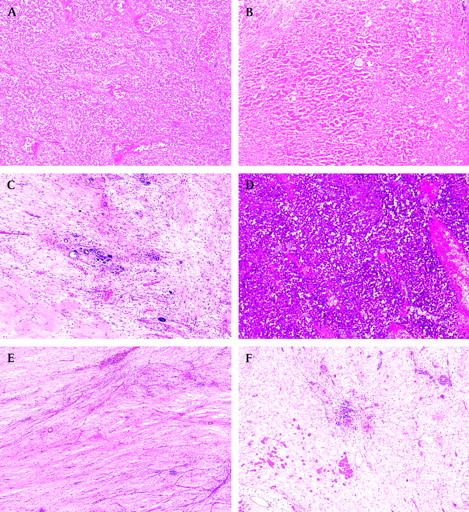
Completely necrotic Wilms' tumour. (A) Non-viable (“ghost”) blastemal cells separated by septa. (B) Non-viable epithelial (tubular) structures clearly identifying a tumour as a Wilms' tumour. (C) A small group of tubules surrounded by non-viable tumour—they are regarded as nephrogenic remnants and should be ignored for tumour's subtyping. (D) “Dying” blastemal cells with still vaguely recognisable nuclear morphology—should be regarded as non-viable tumour and not taken into account for tumour's subtyping. (E) Hypocellular tumour stroma can be indistinguishable from hypocellular areas with chemotherapy-induced changes. (F) Rare rhabdomyoblasts in a hypocellular stroma indicate it is a genuine, viable tumour stroma.

(A) Anaplastic Wilms' tumour with all three features including atypical mitoses, marked nuclear enlargement and hyperchromasia. (B) Focal anaplasia surrounded by chemotherapy-induced changes, making it easier to spot. (C, D) Pseudoanaplasia in the skeletal muscle.
Staging of pretreated Wilms' tumours
Another problem with preoperative chemotherapy is that it makes staging more difficult. However, the results from both SIOP and NWTS/COG trials show that staging is a major problem, not only in pretreated but also in primarily operated tumours.2 16 One of the reasons is that these tumours are usually large and distort the normal architecture of the kidney and its anatomical structures, such as the renal sinus and capsule.
In order to obtain accurate information about the stage of the tumour, the nephrectomy specimen has to be dealt with as detailed in box 1. The two most important steps are to have a (preferably a photo) block guide (figure 3) and to ink the surface of the specimen. Ideally, at least one whole slice of a tumour should be blocked (figure 3), but additional blocks should be taken (box 2).
Box 1 Handling of the nephrectomy specimen
The specimen should be first weighed, measured and photographed. Areas with suspected ruptures and/or invasion should be inked in different colours from the rest of the specimen. A tumour capsule must not be stripped as it would make determination of growth beyond the capsule impossible.
Perirenal and perihilar lymph nodes should be searched for and sampled separately.
The renal vein, artery and ureter should be identified and sampled near the resection margin.
The surface of the whole specimen should be inked and allowed to dry before opening the specimen. This is a crucial step and should always be done; otherwise it might not be possible to stage the tumour.
The specimen should be opened by a longitudinal incision to reveal the tumour and its relation to the kidney, capsule and renal sinus.
The cut surface should be photographed and its macroscopic appearance recorded. The tumour dimensions should be measured and the percentage of necrosis assessed.
Samples of fresh tumour and normal renal tissue should be taken for tumour banking and molecular studies.
The specimen should be fixed in 4% buffered formalin for 24–48 hours, according to the usual procedure of the laboratory. Several additional cuts can be made parallel to the initial cut to divide the specimen into “slabs” for better fixation.
After fixation, blocks should be taken according to the recommended protocol (box 2).

A photo block guide clearly showing where the blocks have been taken from, making accurate staging possible (courtesy of Dr GK Kokai, Royal Liverpool Children's Hospital Alder Hay, Liverpool, UK).
Box 2 Blocks selection from nephrectomy for renal tumours
After fixation, at least one slice of the tumour should be sampled and carefully recorded (figure 3).
In addition, the following blocks should be taken and recorded:
Areas of doubtful resection, as marked by the surgeon or pathologist.
Sinus lymph nodes when present.
Other lymph nodes.
Renal sinus, ureter and sinus vessels. The renal vein should be scrutinised for evidence of tumour thrombus; if present, it is critical to assess whether it is completely resected.
Each nodule away from the main mass (in multifocal tumours).
Tumour–kidney interface.
Tumour–kidney capsule.
Areas of the capsule that are suspected of being invaded by the tumour.
Areas of perirenal fat suspected for tumour infiltration (important for assessment whether the tumour is completely resected).
Areas of adhesions of the tumour to surrounding tissues.
At least two blocks of the normal kidney, and blocks from abnormal looking areas in the remaining renal tissue.
Table 3 shows the staging criteria followed in the SIOP WT 2001 Trial. It is important to remember that the purpose of giving preoperative chemotherapy is to induce a more favourable stage distribution by down-staging tumours. For that reason, staging is based on objective histological findings at the time of nephrectomy rather than on assumptions of what a pathologist thinks had been there before preoperative chemotherapy was given. For example, if there is a viable tumour outside of the kidney, in the perirenal fat, but it is covered by a pseudocapsule (which is likely to be formed due to preoperative chemotherapy), the tumour is still regarded as stage I (figure 4A).
SIOP WT 2001 staging criteria for renal tumours of childhood
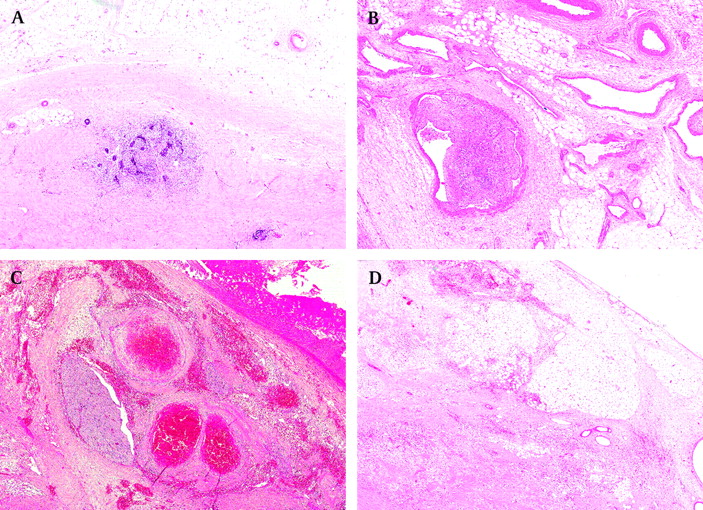
Stage I tumours. (A) Viable tumour at the same level as perirenal fat, suggesting the tumour was extra-renal, but now covered by a pseudocapsule, hence, still stage I. (B) Non-viable tumour thrombus in the renal sinus vessel. (C) Chemotherapy-induced changes in the renal sinus (note a large nerve, confirming it is the sinus). (D) Chemotherapy-induced changes in the perirenal fat are not regarded as a criterion for stage II.
In the current SIOP WT 2001 trial, chemotherapy-induced changes are not taken into account for staging purposes if present in the perirenal fat or renal sinus (either its soft tissues or vessels) (figure 4).18 However, if present at the resection margins in the perirenal fat, in the renal vein or inferior vena cava thrombi, or in lymph nodes (figure 5), they are regarded as a criterion for stage III.
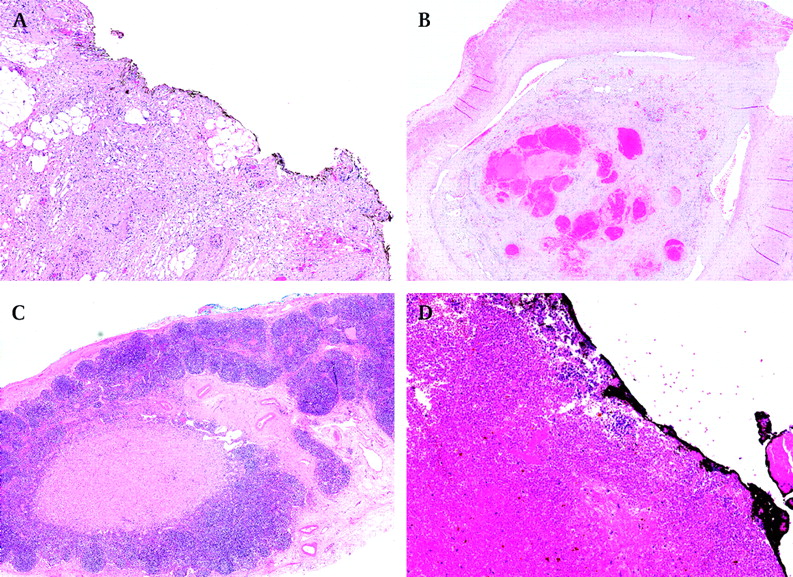
Stage III tumours. (A) Chemotherapy-induced changes at the inked resection margin. (B) Non-viable tumour thrombus at the resection margin of the renal vein. (C) A part of a lymph node is replaced with non-viable tumour. (D) Non-viable blastema at the inked resection margin.
Some staging problems/dilemmas are as follows:
Viable tumour is present around large blood vessels but no other structures of the renal sinus are obvious. WTs often contain large intra-tumoural vessels and they should not be confused with the renal sinus vessels. The most reliable criterion for identifying the renal sinus is the presence of nerves as they are never present in the tumour (figure 4C).
There is no clearly defined (pseudo)capsule and fibrosis is present at the resection margin(s). This should not be regarded as a criterion for stage III—fibrosis may be induced by preoperative chemotherapy, but in order to declare that there is non-viable tumour at resection margins, one should see either “ghost” tumour elements or foamy macrophages (figure 5A,D).
There is clinical information on the form stating that there was a pre-chemotherapy tumour rupture, but there is no evidence of that at nephrectomy. The tumour should be staged objectively, according to pathological findings at nephrectomy; it is the treating paediatric oncologist's decision to assign a treatment stage which may not be based on pathological findings alone.
There are mesenchymal thrombi in the renal sinus vessels but it is not clear whether or not they are viable. It may be, indeed, difficult to distinguish between non-viable and viable stromal thrombi. If no blastema, tubules, rhabdomyoblasts or smooth muscle differentiation are present, one should compare the thrombi with the stromal component of the tumour—if identical, they should be regarded as viable (figure 6).
What changes in a lymph node should be regarded as non-viable metastases? Only if part of a lymph node is replaced with non-viable tumour (usually in a form of foamy macrophages), should it be regarded as evidence of pre-existing tumour and staged as stage III. Massive accumulation of foamy macrophages in the peripheral sinus or within the node, without destruction of normal architecture, is not uncommon but should not upstage a tumour.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Viable tumour is always taken into account for staging. (A) Stage II: viable tumour thrombus in a renal sinus vessel. (B) Stage II: viable stromal thrombus showing similar features as the main tumour. (C) Stage III: viable tumour present at the inked resection margins. (D) Stage III: viable lymph node metastasis.
Finally, the pathology report should be crystal clear in its conclusions, which have to include tumour type, risk group, stage and reason for the stage. For example, a stage III Wilms' tumour should be reported as follows: “Wilms' tumour, regressive type, intermediate risk tumour, stage III (due to lymph node metastases—non-viable tumour)”. A stage II Wilms' tumour should be reported as follows: “Wilms' tumour, mixed type, intermediate risk tumour, stage II (due to renal sinus infiltration—viable tumour)”.
Data from previous trials show that in around 20% of renal tumours there were diagnostic and staging discrepancies between the institutional pathologists and central pathology review.2 The current policy for the UK SIOP WT 2001 trial is that all renal tumours should be submitted for rapid central pathology review, which may allow for changes in management if necessary (box 3).19 The Royal College of Pathologists, in its recently updated guidelines, supports this view, stating that “diagnostic material from patients in a trial in which central review of the specimen is mandated in the trial protocol should be dispatched on request. If this is not done, the referring hospital will be acting against the wishes of the patient who will have given informed consent at entry into the trial, including consent for the stipulated handling of their specimens”.20
Box 3 UK SIOP WT 2001 trial—standard operating procedure for central pathology review
All renal tumours should be submitted for rapid central pathology review with no delay.
Submitted cases should include a full set of H&E slides, (photo) block guide and the institutional pathologist's report (provisional or final).
A set of slides for central pathology review should be prepared at the same time as the set for the institutional pathologist.
The panel/central pathology reviewer will inform the institutional pathologist about their opinion within 24–48 hours of admission of the material.
Take-home messages
Accurate diagnosis, subtyping and staging of nephroblastoma is critical for its correct treatment; it can be achieved through a close collaboration of the institutional pathologists and a rapid central pathology review.
Preoperative chemotherapy alters histological and staging features, and a diagnostic pathologist has to be familiar with these when dealing with the tumour.
The assessment of chemotherapy-induced changes and viable tumour components is the basis of histological subtyping.
The assessment is possible only if the tumour is adequately sampled.
Non-viable tumour present in the renal sinus (both soft tissues and vessels) and perirenal fat does not upstage a tumour from stage I to stage II.
Non-viable tumour present at the resection margins and in a lymph node upstages a tumour in stage III.
The correct staging is only possible if the specimen is inked and there is a block guide.
Acknowledgments
The authors thank the many institutional pathologists who have over the years provided the cases on which the SIOP trials and studies are based, as well as many surgeons, paediatricians, radiation oncologists and other health professionals who managed the children entered on the SIOP trials and studies. We also thank Drs E Lazda, D Brasanac and S Popov for their critical comments on the manuscript.
References
Footnotes
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.