Article Text

Abstract
Aims The overall aim of the new national newborn programme is to identify infants at risk of sickle cell disease to allow early detection and to minimise deaths and complications.
Methods Universal screening for sickle cell disease was introduced in England between September 2003 and July 2006. The 13 newborn laboratories each screen between 25 000 and 110 000 babies a year using the existing dried bloodspot cards. The specified conditions to be screened for include sickle cell anaemia (Hb SS), Hb SC disease, Hb S/β thalassaemia, Hb S/DPunjab and Hb S/OArab. Data are reported on screening results by ethnic group and geographical area.
Results The prevalence of screen positive results across England is 1:2000. There is a 25-fold variation by geographical area. African babies make up 61% of all screen positive results despite representing only 4% of total births. Combined carrier rates vary widely by ethnicity, from 1.85 per 1000 (1:540) in ‘White British’ to 145 per 1000 (1:7) in ‘African’ babies. Refusal rates for screening show variation by ethnicity.
Conclusions These results provide useful information both about the frequency of these conditions and the carrier state and their geographic and ethnic distribution across England. This can be used to refine counselling information and are also useful to target and plan services and public information.
- Sickle cell
- newborn screening
- haemoglobinopathies
- sickle cell carrier
- sickle cell prevalence
- newborn biochemical screening
- sickle cell disease
- haemoglobinopathy
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Sickle cell
- newborn screening
- haemoglobinopathies
- sickle cell carrier
- sickle cell prevalence
- newborn biochemical screening
- sickle cell disease
- haemoglobinopathy
Aims
The overall aim of the recently introduced national newborn programme is to identify infants at risk of sickle cell disease to allow early detection and to minimise deaths and complications through early treatment and care. Screening is now offered to all babies in England irrespective of ethnicity at 5–8 days of age as part of the pre-existing newborn dried blood-spot screening programme; implementation is planned for Scotland in 2010. We report the frequency of suspected disease rates for different parts of the country and for the first time we report newborn carrier data broken down by ethnicity using the categories recorded on the dried bloodspot (Guthrie) card. This is important information for those who have to counsel such families. The reported carrier rate for England as a whole is 15/1000. The frequency of diagnosis of sickle cell disease suggests that it should be given a higher priority in medical and nursing education and in NHS service planning and provision. The scale of need for genetic counselling arising from this programme is also emphasised.
Methods
Screening for sickle cell disease was introduced in England between September 2003 and July 2006, building on the existing patchy screening arrangements which covered about 15% of England (in parts of London and the West Midlands).
The policy adopted by the National Screening Committee (NSC) was to introduce universal resident based screening of all infants using the bloodspot card and to discontinue catchment based cord blood samples.1 The newborn laboratory service providing the screening in England now consists of 13 centralised biochemistry newborn screening laboratories—each screening between 25 000 and 110 000 babies a year. For sickle cell screening, second line testing of all positive results is performed, either within the screening laboratory, or by a specialist haematology laboratory within the same hospital trust or elsewhere. Two methods of analysis are applied to all screen-positive results obtained from dried blood spot samples to ensure high specificity: either high performance liquid chromatography or iso-electric focussing (further details are included in our laboratory handbook).2 In addition to screening for sickle cell disease, these biochemistry laboratories perform newborn screening for phenylketonuria, congenital hypothyroidism, cystic fibrosis and medium chain acyl CoA dehydrogenase (MCADD).3
The screening programme specifies the conditions to be screened for and recommends specific methods to be used.2 The conditions screened for because of the potential benefit are: sickle cell anaemia (Hb SS), Hb SC disease, Hb S/β thalassaemia, Hb S/DPunjab and Hb S/OArab. Hb S/hereditary persistence of foetal haemoglobin is also included as it needs to be distinguished from Hb SS and S/β or δβ thalassaemia although it is not in itself a condition for which there is proven benefit in screening. Currently the UK NSC does not support screening for β thalassaemia.
A detailed implementation plan was developed which included training of staff responsible for taking samples (predominantly midwives) and providing laboratory set-up costs, development of materials for parents, recruitment of counsellors and funding of laboratories as described elsewhere.4 5 Standards for the linked newborn and antenatal programme6 and for the overall bloodspot programme3 are available in various publications.
The NSC supported the recommendation by the programme that, in line with pre-existing practice in areas already undertaking newborn screening, carriers of the main haemoglobins including S, C, D, E and others should be reported to parents. This was a controversial area with many clinical geneticists considering that this information should be withheld, but the debate has helped to move thinking forward on this issue. The UK decision is in line with the recent review undertaken in the USA and takes account of the fact that the aim of the programme is to detect infants affected by conditions for which there is good evidence of benefit.
Screening for carriers is not the primary aim of the programme, but these are detected using our chosen technology. In the UK it was not considered ethical to withhold this information if parents wished to have it.7 8 Carriers of the common haemoglobin variants (S, C, D and E) are therefore reported to parents with specific supporting materials aimed of parents with an infant rather than adults considering a pregnancy.9 Counselling is offered to ensure parents understand that their child is healthy and the difference between carrier state and disease, the distinction between the S carrier status (which is clinically important to know) and other carrier statuses (which are unlikely to be clinically important but have genetic relevance).9 Unlike antenatal screening, newborn screening methods do not reliably identify all thalassaemia carriers although parents who are carriers often want to know this information. Recently published work has suggested that some β thalassaemia carriers may be detected by their low levels of Hb A at birth.10
Demographic data are recorded on the blood spot card by the midwife who takes the sample, including ethnic data selected from a table printed on the back of the card. This ethnic data field is not completed in all cases and has not been available from one laboratory for the period reported here.
Data are collected from all laboratories, checked and discussed with the providing laboratory to keep inconsistencies to a minimum. The data are used to assist in planning counselling and clinical services and to inform antenatal prevalence estimates and local screening policy and service planning. The time period of the data collections is the two financial years of 2005/06 and 2006/2007.
Results
The prevalence of screening results indicating likely sickle cell disease (screen positive results) varies widely (table 1). It ranges from 3 per 1000 in South East London (ie, about 1:330 babies have a screen positive result) to 0.12 per 1000 in Cumbria and Lancashire—a 25-fold variation in prevalence. Within London the prevalence is highest in South East London at 3/1000 and lowest in North West London at 1/1000. Other areas with higher rates of 0.4–0.5/1000 include Leicester and Northants, Bedfordshire, Birmingham, Essex and Greater Manchester. Overall babies recorded as Black African make up 61% of all the sickle cell disease suspected results despite representing only 4% of total births. Overall screen positive disease prevalence rate is 1:2000 for England.5
Rates of significant conditions* by strategic health authorities†: April 2005 to March 2007
Figure 1 shows that combined carrier rates also vary widely by ethnicity from 1.85 per 1000 (1:540) in those babies recorded as ‘White British’ to 145 per 1000 (1:7) for ‘Black African’ babies.

Carrier rates by ethnic category: April 2005 to March 2007.
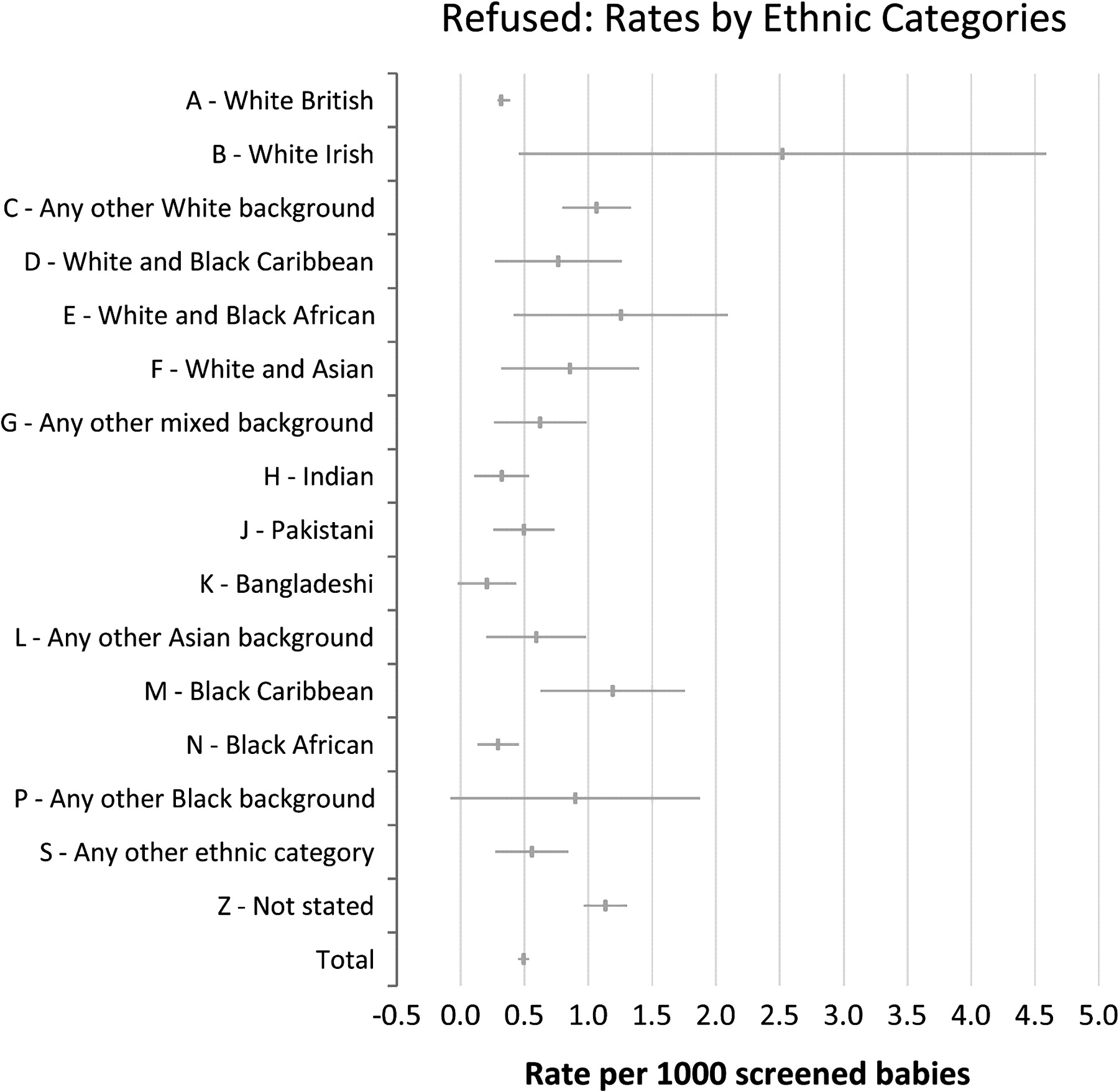
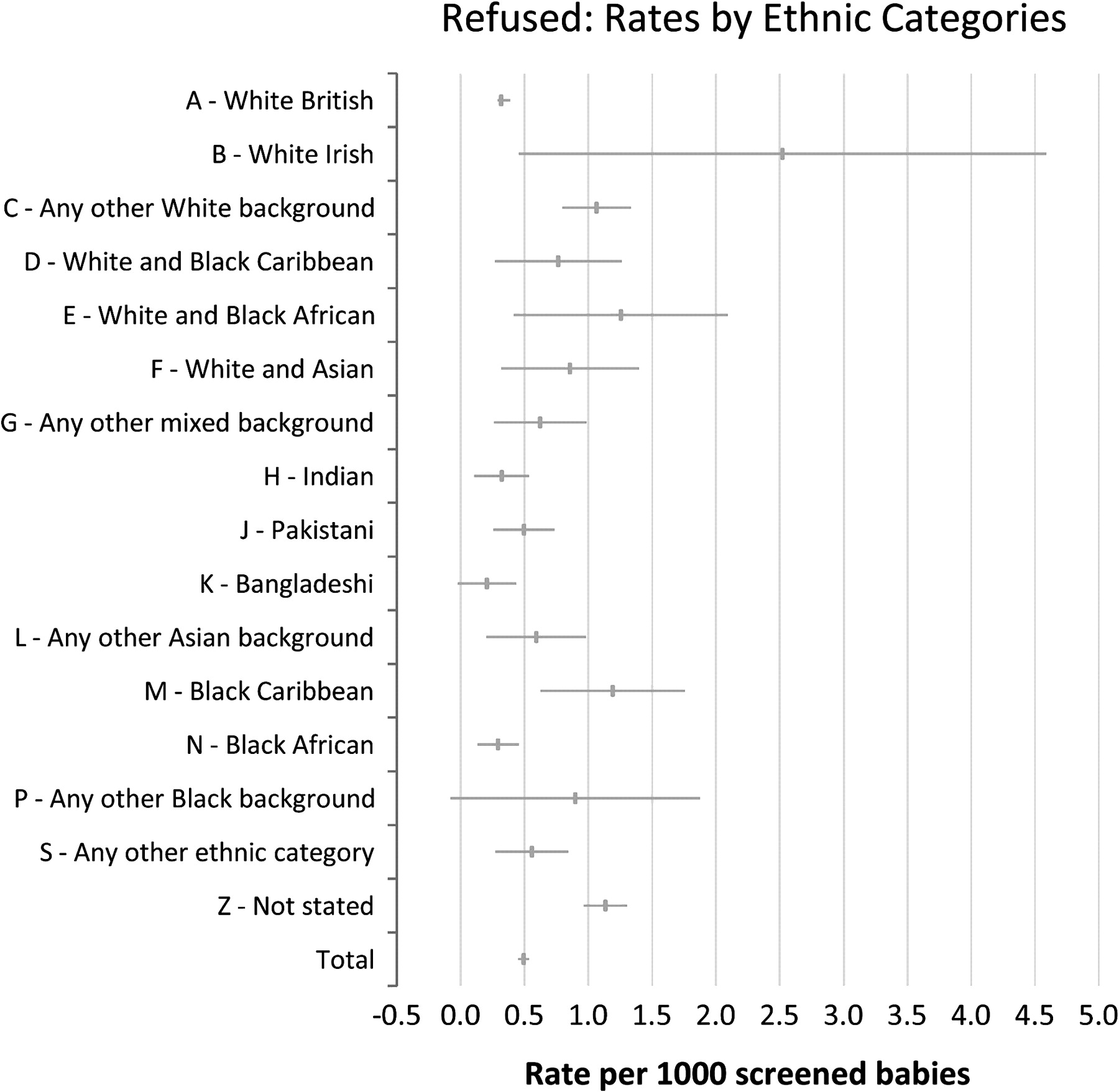
Figure 2, showing refusal rates for screening, shows that several populations such as the ‘any other white background’ (1:940), ‘Black Caribbean’ (1:840) and ‘not stated’ (1:880) populations appear to have a significantly higher rate of refusal of screening than the ‘White British’ (1:3150) category with CIs not overlapping the ‘White British’ category. Overall about 300 refusals are documented each year, and for these cases a blank card is sent to the laboratory to ensure that this information is documented on all records so that a test may still be performed if clinical suspicion is raised or if the child subsequently becomes ill. Almost exclusively refusals are for screening for all five conditions and not specifically a refusal for sickle cell disease screening. To date, to our knowledge, none of these refusals have been babies already identified by antenatal screening as at high risk of being affected. For infants at a 1:4 or higher risk the clinical policy is to offer liquid blood sample testing in advance of the bloodspot screen which is still offered (see page 4 of the programme laboratory handbook).2
{kind=link}
{kind=link}
Refusal rates per 1000 babies screened: April 2005 to March 2007.
Table 2 shows that different ethnic groups have different patterns of carrier rates, with haemoglobin D being predominantly seen in the Indian and Pakistani populations and haemoglobin E seen in the Bangladeshi population.
Carrier rates by ethnic category* and Hb type: April 2005 to March 2007
Conclusion
These results from the newborn screening programme provide useful objective information, both about the frequency of these conditions and the carrier state and their variable geographic and ethnic distribution across England. They give ratios of conditions and carrier frequencies by ethnic group which can be used by the programme to refine counselling information for individuals and couples as recently recommended by Kai et al.11 They are also useful to target and plan services and public information.
The limitations of the data are that it is based on ethnicity data ascertained by midwives and recorded on the bloodspot card rather than on a detailed family origin questionnaire, and also that ethnic data were not recorded in about 13% of samples. Despite this, the overall pattern and distribution of disease and carrier rates expected is clearly shown. A second limitation of the data is that these are screening and not diagnostic results, but as reported elsewhere the screening methods used are highly specific and sensitive, and results are unlikely to be significantly changed when confirmatory tests are completed.5
None of these limitations are likely to materially affect the main findings of the general prevalence of the condition—now about 1:2000 affected births in England: it is as common as cystic fibrosis. The significant contribution that the Black African, rather than the African-Caribbean population makes to the disease is of note. The data also show the significant burden that these conditions are likely to place on the London NHS.
These objective figures should replace previous estimates of the scale of these conditions which are now out of date. They suggest that the management and care of haemoglobinopathies as a long term condition needs to move from its ‘orphan’ status into the mainstream of the NHS commissioning and clinical agenda as an important addition to the inequalities in healthcare agenda. The huge variation in the condition by ethnicity shown by these figures arguably explains why the condition has not received the attention it merits as the groups affected are often marginalised within UK society. A welcome recent development which may be helping to raise the profile of these conditions in the NHS is the establishment of an All Party Parliamentary group.
The other issue that these figures raise is the frequency of the carrier state and the ongoing challenge to record such information accurately, in primary care records, so that it is available when needed and repeat testing is avoided. In the case of S carrier status, this is important information for parents and children as sickling does occur, albeit rarely, under stress of deoxygenation such as during surgery, at high altitude and during extreme physical activity.12 13 This information is not solely relevant for reproductive reasons but also for clinical reasons, and in a child this latter point is the important issue.
We hope that by presenting objective information, which should be read alongside other information, the clinical community responsible for education of future clinicians and for developing services will act to support the obvious clinical needs shown, and will ensure that genetic issues are considered alongside clinical needs.14
Take-home messages
In England, sickle cell disease is considerably more common than usually quoted, with a birth prevalence of 1:2000 (more common than cystic fibrosis=1:2500), and an S carrier rate of almost 1% in newborn babies.
There is wide (25-fold) geographical variation in the frequency of sickle cell disease. Most affected babies are in London and other large urban centres, but sickle cell disease is occurring in all parts of England, including areas where previously the disease was unreported and thought to be ‘not a problem here’.
For the first time reliable carrier rates by reported ethnicity give objective information by ethnic group to help those who counsel individuals and couples about their risk of sickle cell conditions. This includes the fact that 1:800 babies reported as ‘White British’ is a carrier of the S, C, D or E gene.
Collecting this ethnicity data has also been very valuable in showing variation by ethnicity in refusal rates, and this should continue to be monitored.
Acknowledgments
We acknowledge the programme steering group, including the chair, the Right Honourable and Most Reverend Dr John Sentamu, Archbishop of York; the regional teams that coordinated the implementation and the various laboratory subgroups; and the programme centre team, including Dr Sandra Anglin, Cynthia Gill and Sir Muir Gray for support in planning and policy development at the start of the programme. We also acknowledge the laboratories that perform screening and the individuals named who contributed screening data from those laboratories to the programme: Dr Kate Hall, Birmingham Children's Hospital; Dr Helena Kemp and Dr Jacky James, Southmead Hospital, Bristol; Dr Jacqui Calvin, Addenbrooke's Hospital, Cambridge; Dr Mary Clarke, Epsom and St Helier NHS Trust; Mrs Lisa Farar, St James's University Hospital, Leeds; Mr Paul Walsh and Dr R Keenan, Royal Liverpool Children's Hospital; Dr Chris Lambert, King's College Hospital, London; Ms Mandy Pickersgill, Royal Manchester Children's Hospital, Manchester; Ms Lisa Thomas, Royal Victoria Infirmary, Newcastle-upon-Tyne; Dr Shirley Henderson, John Radcliffe Hospital, Oxford; Dr David Sinclair, Queen Alexandra Hospital, Portsmouth; Ms Melanie Downing and Mrs Angela Matthews, Sheffield Children's Hospital, Sheffield; and Dr Steve Kryvovich, Great Ormond Street Hospital, London.
Footnotes
Sources and selection criteria: The data reported are obtained from the annual screening data return that each newborn laboratory provides to the screening programme, now on an annual basis. They were obtained by JH and then checked and collated by the programme office (RL); queries were directed to individual laboratories (contacts acknowledged in this paper) when inconsistencies were identified or problems arose. Not all data were available from all laboratories for all time periods—where this is the case it is indicated in the figures/tables as numbers may vary. The programme does not hold patient identifiable data from this source and does not publish data where there are fewer than five cases in a box.
Funding Funding of the implementation programme and establishment of the programme centre are provided by the Department of Health.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.