Article Text

Abstract
Aims Detecting low-level clinically significant cancer-relevant somatic mutations can be difficult. Several technologies exist for detecting minority mutations. One method is locked nucleic acid (LNA) PCR. In this study, LNA probes were used to enhance the sensitivity for detecting FLT3 D835/I836 tyrosine kinase domain (TKD) mutations, the JAK2 V617F mutation and insertion mutations in the nucleophosmin 1 gene.
Methods PCR was performed with and without LNA probes using DNA known to contain FLT3 D835/I836 TKD, JAK2 V617F and NPM1 mutations. FLT3 D835/I836 TKD mutations were detected following EcoRV restriction enzyme digestion and capillary electrophoresis. The JAK2 V617F mutation was detected by melt-curve analysis. NPM1 insertions were detected by capillary electrophoresis.
Results The detection of FLT3 D835/I836, JAK2 V617F and NPM1 mutations was enhanced approximately 10–50-fold using LNA probes. Rare JAK2 double mutants gave abnormal blocking patterns with the LNA probe.
Conclusions Adding LNA probes to existing assays is a simple way to enhance and confirm the detection of mutations, especially those at low levels.
- Colorectal cancer
- diagnosis
- diagnostics
- gall bladder
- haemato-oncology
- haem-oncology
- molecular genetics
- molecular pathology
- oncogenes
- pancreas
- p53
Statistics from Altmetric.com
- Colorectal cancer
- diagnosis
- diagnostics
- gall bladder
- haemato-oncology
- haem-oncology
- molecular genetics
- molecular pathology
- oncogenes
- pancreas
- p53
Detecting low-level clinically significant somatic mutations in cancer can be difficult if a large excess of wild-type DNA is present. One method to enrich for low-level mutations is locked nucleic acid (LNA) PCR. LNA is a nucleic acid analogue that contains a 2′-O,4′-C-methylene bridge in the ribose moiety. LNA bases can be incorporated into any DNA oligonucleotide, and each LNA base increases by 3–8°C the thermal stability with complementary DNA. When oligonucleotides contain wild-type-specific LNA bases, LNA/DNA hybrids with high thermal stability are formed and wilt-type allele amplification is suppressed. If a mutation is present, the LNA probe does not bind to the mutant allele, and the mutant allele can be preferentially amplified.1 2
In this study, LNA probes were added to assays to detect FLT3 D835/I836 tyrosine kinase domain (TKD), JAK2 V617F and nucleophosmin (NPM1) insertion mutations in order to determine whether the sensitivity of detecting mutant alleles could be improved.
Mutations in the FLT3 TKD occur in approximately 5–11% of acute myeloid leukaemia (AML) patients, but the prognostic significance of these mutations in cytogenetically normal AML is controversial, with differences in favourable3 versus adverse4 outcomes being attributed to low versus high mutant levels, mutation detection methods, treatment, patient populations5 6 and effects of NPM1 and CEBPA mutations.7 Mutations at codons 835 and 836 are the most common TKD mutations and disrupt an EcoRV restriction site, which facilitates simple screening,8 but distinguishing incomplete digestion from low-level mutants can be problematical. Herein, a series of LNA probes were synthesised and one was found to enhance D835/I836 mutation detection.
The JAK2 V617F (1849G>T) point mutation is found in approximately 95% of cases of polycythaemia vera and approximately 50% of cases of essential thrombocythemia and primary myelofibrosis. JAK2 mutation analysis has been endorsed by the WHO for diagnosing these disorders.9 A number of different assays with varying sensitivities to detect this mutation have been described.10 One method is melt-curve analysis, but this can yield difficult-to-interpret ‘shoulder peaks’ when the mutant level is less than 10%.10 Melt-curve analysis, unlike allele-specific PCR or assays that use probes that detect only wild-type or mutant alleles, can, however, detect novel variants that lie underneath the sensor probe and several non-V617F variants have been identified this way.11 12 In the current study, a LNA probe used in a molecular beacon assay13 was added to a melt-curve assay.14 The LNA probe was also evaluated using rare JAK2 double mutants (V617F/C618R and V617F/C618F).12
Exon 12 insertion mutations in the NPM1 gene are found in approximately 25–35% of all cases of AML and 46–64% of karyotypically normal AML.15 The advantages and disadvantages of different methods to detect NPM1 mutations have been reviewed.16 As NPM1 mutations appear stable, they represent a minimal residual disease marker that requires more sensitive assays to detect low mutant levels.15 16 An NPM1 LNA probe used in a melting assay17 was evaluated in a PCR and capillary electrophoresis assay.
Materials and methods
FLT3 D835/I836 TKD mutation detection
PCR primers (Invitrogen, Carlsbad, California, USA) were 5′–FAM-GAAAGATTGCACTCCAGGATAAT–3′ (forward) and 5′–GCCGTATAAAAATAAGTAGGAA–3′ (reverse). LNA probes were from Exiqon (Woburn, Massachusetts, USA), and the sequence of the LNA probe selected for further evaluation was GCTCG+A+G+A+T+ATCATGAG where + refers to locked bases. Each 25 μl PCR reaction contained 12.5 μl 2× Qiagen HotStart Master Mix (Valencia, California, USA), 0.5 μM of forward and reverse primers, approximately 100 ng DNA, and, when indicated, 5 μM LNA probe. PCR was performed in a MJ Research PTC-225 DNA Engine Tetrad using one cycle of 95°C for 3 min followed by 49 cycles of 95°C for 15 s, 55°C for 15 s, and 72°C for 15 s with a final 10 min extension at 72°C and a 4°C hold. PCR products were digested with EcoRV (New England Biolabs, Ipswich, Massachusetts, USA) and analysed by capillary electrophoresis on an ABI 3730 DNA analyser (Life Technologies Corporation, Carlsbad, CA).
JAK2 V617F mutation detection
Melt-curve analysis to detect the JAK2 V617F mutation was identical to that described previously14 except the sensor probe was mutant (617F) (5′–ATGGAGTATGTTTCTGTGG–flourescein–3′) rather than wild-type (617V) specific in order to maximise mutant JAK2 detection because the LNA probe is wild-type specific. The LNA probe (Exiqon) sequence13 was +T+A+T+G+T+G+T+C+T+GT where + refers to locked bases and G is the JAK2 V617F mutation site (1849G>T). When indicated, 0.25 μM of the LNA probe was included. Primers and probes were from TIB Molbiol (Nutley, New Jersey, USA) and Integrated DNA Technologies (Coralville, Iowa, USA). The HEL cell line was obtained from DSMZ (Braunschweig, Germany).
NPM1 mutation detection
PCR primers (Invitrogen) were 5′–FAM-TTTTTCCAGGCTATTCAA–3′ (forward) and 5′–CAGGCATTTTGGACAACACA–3′ (reverse). The LNA probe (Exiqon) sequence17 was: 5′–A*A*+G+ATCT+CT+G+GCA+GT+GG/3SpC3/–3′ where *, + and /3spC3/ refer to phosphorothioate, LNA and C3 spacer, respectively. Each 25 μl PCR reaction contained 12.5 μl 2× Qiagen HotStart Master Mix, 0.5 μM of forward and reverse primers, approximately 100 ng DNA, and, when indicated, 5 μM LNA probe. PCR was performed in a MJ Research PTC-225 DNA Engine Tetrad using one cycle of 95°C for 15 min followed by 44 cycles of 95°C for 30 s, 54°C for 30 s and 72°C for 30 s, with a final 10 min extension at 72°C and a 4°C hold. PCR products were analysed by capillary electrophoresis on an ABI 3730 DNA analyser. The OCI-AML3 cell line was obtained from DSMZ.
Results
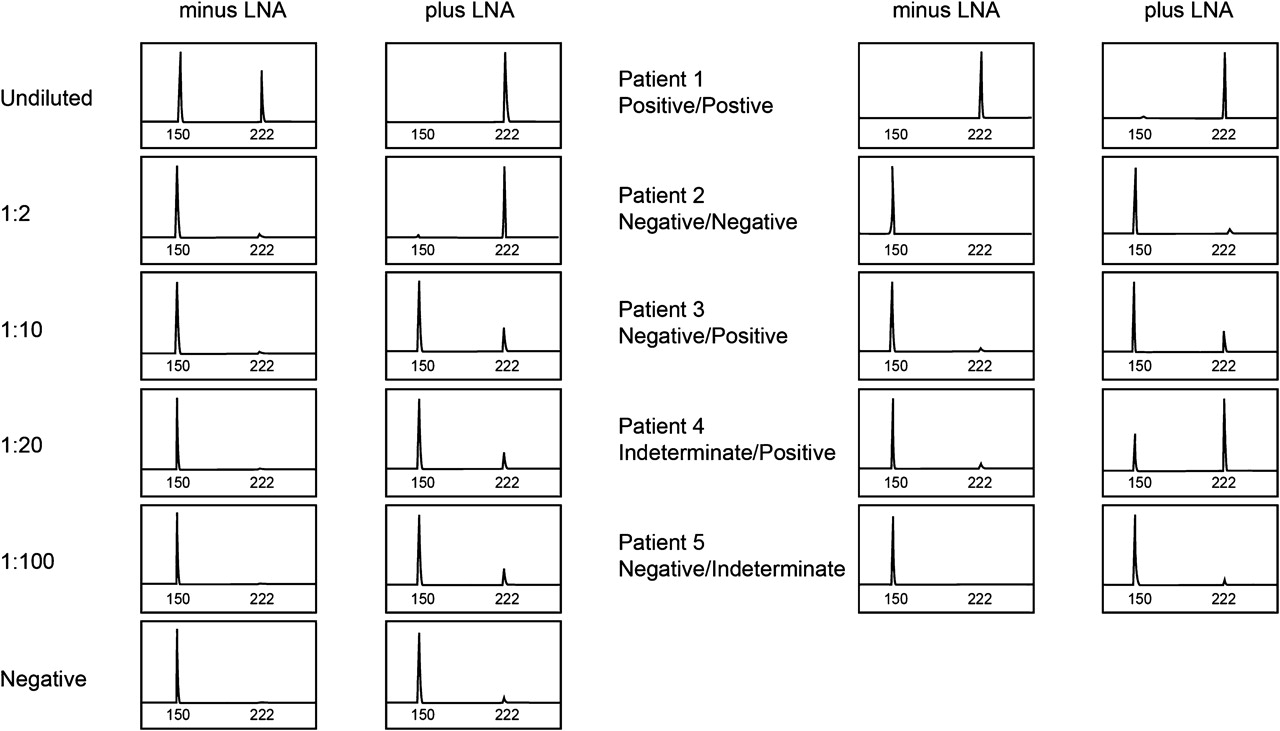
Nine LNA probes complementary to wild-type FLT3, which spanned amino acids 835 and 836 of the TKD and varied in both the positions of locked bases and 5′ and 3′ modifications, were synthesised and screened for their ability to enhance mutant allele detection (data not shown). The most effective LNA probe was evaluated further, and using D835/I836 mutant DNA diluted into wild-type DNA, figure 1 shows that the LNA probe enhanced the detection of the mutant 222 base pair allele from 1:2 (− LNA) to 1:100 (+ LNA). Thirty-six patient samples that had previously been submitted for routine clinical testing were re-tested with and without the LNA probe. Because mutation negative samples consistently showed a small amount of undigested product, which was greater in the presence of the LNA probe than in its absence, samples were scored positive with the LNA probe if mutant/wild-type peak height ratios were greater than a 1:100 control and indeterminate if ratios were in between 1:100 and mutation negative controls. Of 36 samples tested in the absence and presence of the LNA probe, 13 were positive/positive, 10 were negative/negative, six were negative/positive, three were indeterminate/positive and four were negative/indeterminate. Figure 1 also shows representative samples in the absence/presence of the LNA probe from patients who were positive/positive (patient 1), negative/negative (patient 2), negative/positive (patient 3), indeterminate/positive (patient 4) and negative/indeterminate (patient 5). These results demonstrate that adding the LNA probe enhances the detection of the mutant allele and can help resolve differentiating low-level mutants from incomplete restriction enzyme digestion.

Left: D835/I836 positive DNA was serially diluted at the indicated levels into normal DNA and subjected to PCR in the absence and presence of 5 μM locked nucleic acid (LNA) probe. Undiluted D835/I836 positive and negative DNA were also run. Right: examples of patient samples that were positive/positive, negative/negative, negative/positive, indeterminate/positive and negative/indeterminate in the absence and presence of 5 μM LNA probe are shown. After PCR, products were digested with EcoRV and analysed by capillary electrophoresis. D835/I836 negative samples show a 150 bp fragment. When a mutation is present, an additional EcoRV resistant fragment at 222 bp is seen.
In figure 2, the indicated ratios of JAK2 V617F mutant (HEL cell) to wild-type DNA were prepared and mutation detection was performed using melt-curve analysis14 with a mutant sensor probe in the presence and absence of the LNA probe. In the absence of the LNA probe, the 617F allele was slightly detected at mutant/wild-type ratios of 5/95 (panel B) and not at 1/99 (panel C). However, in the presence of the LNA probe, the 617F allele was easily detected at mutant/wild-type ratios of 5/95 (panel B), 1/99 (panel C), 0.5/99/5 (panel D) and slightly at 0.1/99.9 (panel E). These results show that the LNA probe resolved problems associated with interpreting shoulder peaks in low-level mutants10 by magnifying the mutant peak. Six patient samples suspicious for low levels of the JAK2 V617F mutation in the absence of the LNA probe were re-tested with the LNA probe. All samples showed complete blockage of amplification of the wild-type allele and enhanced detection of the mutant allele. These six samples were also positive using the Ipsogen MutaScreen Kit (data not shown).

Locked nucleic acid (LNA) probe enhances the sensitivity of mutant JAK2 detection and completely blocks amplification of wild-type (WT) DNA. In panels A-F, the indicated rations of mutant (HEL cell) to WT DNA were prepared. JAK2 V617F mutation detection was performed using melt-curve analysis in the presence and absence of 0.25 mM LNA probe.
Two rare and previously sequenced confirmed JAK2 double mutants, which were originally identified because of atypical melt-curves using a wild-type-specific sensor probe,12 were re-tested using a mutant-specific sensor probe in the absence and presence of the LNA probe. The V617F/C618R ‘homozygous’ double mutant from a patient with polycythaemia vera gave an atypical melting curve (Tm ∼48°C) (figure 3A) which failed to block using the LNA probe (figure 3B). Interestingly, a low-level V617F/C618F double mutant in a patient being treated with Hydrea for essential thrombocythaemia that gave an atypical melting curve using a wild-type-specific probe12 gave a melting curve virtually identical to wild-type JAK2 (Tm ∼50°C) (figure 3A). However, this mutant failed to block completely in the presence of the LNA probe (figure 3B) so this rare JAK2 variant would not have been incorrectly genotyped as wild type because failure to block with the LNA probe would trigger sequence analysis.

Melt curve analysis of JAK2 double mutants in the absence (A) and presence (B) of 0.25 μM locked nucleic acid (LNA). The LNA probe blocks amplification of the wild-type (WT) allele (Tm ∼50°C) using 100% WT JAK2 and a 50/50 mixture of HEL cell DNA and WT DNA (mixture mutant/WT). The LNA probe does not block amplification of the mutant allele (Tm ∼58.5°C) using 100% mutant (HEL cell) DNA or mixture mutant/WT DNA. The V617F/C618R double mutant gives an atypical melting curve, which fails to block with the LNA probe. The V617F/C618F double mutant gives a melting curve indistinguishable from WT JAK2 but fails to block with the LNA probe.
Figure 4 shows NPM1 mutation detection in the absence and presence of the LNA probe using the indicated ratios of OCI-AML3 cell (mutant) to wild-type DNA. When a NPM1 mutation is present, one normal 215 bp and one mutant 219 bp product are seen. The LNA probe blocks amplification of the wild-type 215 bp allele and enhances detection of the mutant 219 bp allele. The mutant 219 bp allele is detected minimally using a mixture of 10% mutant/90% wild-type DNA and is not seen using 5% mutant/95% wild-type or 1% mutant/99% wild-type DNA. The LNA probe enhances detection of the mutant allele to 1% mutant/99% wild-type levels.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Locked nucleic acid (LNA) probe enhances the sensitivity of NPM1 insertion mutation detection. The indicated ratios of mutant (OCI-AML3 cell) to wild-type (WT) DNA were prepared. NPM1 insertion mutation detection was performed by PCR in the presence and absence of 5 μM LNA probe followed by capillary electrophoresis. The 215 bp peak is the WT allele and the 219 bp peak is the mutant allele with a 4 bp insertion.
To validate the NPM1 assay, 24 patient samples that had been tested at an external laboratory were tested with and without the LNA probe. Agreement was seen in 23/24 samples. The one discordant sample that was negative at the external laboratory was positive with the LNA probe and barely detected without the LNA probe. This sample was subsequently tested at a different reference laboratory and was positive (data not shown). The discordant specimen was from a patient with recurrent/persistent AML; an initial diagnostic specimen from this patient was NPM1 positive. It is likely that the discrepant sample was below the sensitivity of the initial external laboratory.
Discussion
In this study, we demonstrate that adding LNA probes to three separate assays to detect FLT3 D835/I836 TKD, JAK2 V617F and NPM1 mutations enhances mutation detection 10–50 fold. The LNA probes used for enhancing JAK2 V617F13 and NPM117 had been used previously in molecular beacon and melting assays, respectively. In the current study, we show that these LNA probes are effective when used with different methodologies and demonstrate the versatility of these probes. Although running samples with and without LNA probes increases testing costs, it has the advantage of unambiguously confirming positive samples and eliminating the need to repeat samples that may appear equivocal or indeterminate.
We also report a novel FLT3 D835/I836 LNA probe that was especially useful in differentiating incomplete restriction enzyme digestion from low-level mutants. This LNA probe also identified 9/36 (25%) samples that were FLT3 D835/I836 mutation positive only in the presence of the LNA probe. Therefore, the prevalence of D835/I836 TKD mutations, especially low-level mutants, is likely to be higher than originally reported. Analogous findings were recently described by Arcila et al,18 who found a 17% higher detection rate for the endothelial growth factor receptor T790M mutation using a LNA-based method. As low-level FLT3 TKD mutations may be associated with higher relapse rates than high levels of mutations,6 standardising testing methods for mutant level determination is required in order to determine whether these mutations correlate with outcome. Nevertheless, adding the LNA probe is a simple way to confirm and enhance FLT3 D835/I836 TKD mutant detection, especially those at low levels.
For JAK2 V617F mutation detection, the simple melt-curve assay coupled with a LNA probe described herein enhanced the sensitivity of mutant JAK2 detection 10-fold and eliminated the problem of difficult-to-interpret ‘shoulder peaks’ at mutant levels less than 10%.10 In addition, the LNA probes identified rare JAK2 double mutants by generating abnormal blocking patterns in samples that gave both atypical and typical melting curves. Novel variants have been identified in melting assays by generating atypical melt curves.19 In the current study, the melt-curve assay did not identify the V617/C618F variant because the melting curve generated looked like a wild-type sample. However, this rare double mutant was identified because the LNA probe did not completely block amplification of the wild-type allele. It should be noted, however, that because Ipsogen has a patent on V617F mutation analysis,20 melt-curve analysis coupled with a LNA probe would be limited to research use only.
For NPM1, the LNA probe enhanced the sensitivity for detecting insertion mutations approximately 10-fold and we routinely analyse samples submitted for clinical testing with and without the LNA probe. As NPM1 mutations appear stable and thus can be considered a minimal residual disease marker,15 16 including the LNA probe is a simple and effective way to enhance minimal residual disease detection.
In conclusion, new technologies are allowing for the detection of minority DNA mutations, and abilities to enrich for low-level mutants are becoming increasingly important in clinical and diagnostic applications, especially in those related to cancer detection and targeted therapy. Precise sensitivity requirements for mutation detection may not be a ‘one fits all solution’; too sensitive assays may detect minor mutant populations that could potentially deny patients with predominantly non-mutant tumour treatment benefits or too sensitive assays may detect low levels of mutants found in healthy individuals. Conversely, highly sensitive assays are desirable for minimal residual disease detection following treatment, for detecting rare circulating tumour cells and in solid tumours, such as metastatic lesions, in which tumour percentages may be low. Nevertheless, it will be interesting to see whether LNA probes can be combined with other novel approaches such as COLD-PCR or digital PCR to enrich further for the detection of low-level somatic mutations.
Take-home messages
Detecting low-level clinically significant cancer-relevant somatic mutations can be difficult and LNA PCR can be used to enhance the detection of minority mutations.
LNA PCR enhanced the sensitivity for detecting FLT3 D835/I836 TKD mutations, the JAK2 V617F mutation and insertion mutations in the nucleophosmin 1 gene approximately 10–50-fold. Rare JAK2 double mutants were also identified using LNA PCR.
Adding LNA probes to both existing and novel assays is a simple way to enhance and confirm the detection of mutations, especially those at low levels.
References
Footnotes
Funding This study was funded by the Research Program Committee Awards Program at the Cleveland Clinic Foundation.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.