Article Text

Abstract
Iron functions Iron is an essential micronutrient, as it is required for satisfactory erythropoietic function, oxidative metabolism and cellular immune response.
Iron physiology Absorption of dietary iron (1–2 mg/day) is tightly regulated and just balanced against iron loss because there are no active iron excretory mechanisms. Dietary iron is found in haem (10%) and non-haem (ionic, 90%) forms, and their absorption occurs at the apical surface of duodenal enterocytes via different mechanisms. Iron is exported by ferroportin 1 (the only putative iron exporter) across the basolateral membrane of the enterocyte into the circulation (absorbed iron), where it binds to transferrin and is transported to sites of use and storage. Transferrin-bound iron enters target cells—mainly erythroid cells, but also immune and hepatic cells—via receptor-mediated endocytosis. Senescent erythrocytes are phagocytosed by reticuloendothelial system macrophages, haem is metabolised by haem oxygenase, and the released iron is stored as ferritin. Iron will be later exported from macrophages to transferrin. This internal turnover of iron is essential to meet the requirements of erythropoiesis (20–30 mg/day). As transferrin becomes saturated in iron-overload states, excess iron is transported to the liver, the other main storage organ for iron, carrying the risk of free radical formation and tissue damage.
Regulation of iron homoeostasis Hepcidin, synthesised by hepatocytes in response to iron concentrations, inflammation, hypoxia and erythropoiesis, is the main iron-regulatory hormone. It binds ferroportin on enterocytes, macrophages and hepatocytes triggering its internalisation and lysosomal degradation. Inappropriate hepcidin secretion may lead to either iron deficiency or iron overload.
- Diagnosis
- haemochromatosis
- hematopoesis
- inflammation
- metabolism
Statistics from Altmetric.com
Introduction
Iron is an essential micronutrient, as it is required for satisfactory erythropoietic function, oxidative metabolism and cellular immune response. For a 70 kg man, total body iron is about 3500 mg (50 mg/kg body weight). Most of the iron in the body is distributed in red blood cell haemoglobin (65%; 2.300 mg). Approximately 10% is found in muscle fibres (in myoglobin) and other tissues (in enzymes and cytochromes) (350 mg). The remaining body iron is stored in the liver (200 mg), macrophages of the reticuloendothelial system (RES) (500 mg) and bone marrow (150 mg).
On the other hand, the body has no active means of excreting iron, and thus regulation of absorption of dietary iron from the duodenum plays a critical role in iron homoeostasis.1 This is extremely important as iron is essential for cellular metabolism and aerobic respiration, and cellular iron overload leads to toxicity and cell death via free radical formation and lipid peroxidation. Thus, iron homoeostasis requires tight regulation.2–4
In this paper, we will review the main pathways of iron metabolism and their regulation (part I), whereas the causes of iron deficiency and iron overload, and the different laboratory tests to establish a correct diagnosis of iron overload, iron deficiency and anaemia, and the indications, advantages and side effects of the different options for treating iron overload and iron deficiency will be discussed in the second paper (part II)
Main pathways of iron homoeostasis
Iron absorption
The normal Western diet contains 15–20 mg iron in haem (10%) and non-haem (ionic, 90%) forms, of which 1–2 mg are absorbed daily mostly at the duodenum. Iron absorption is balanced against iron loss through sloughed intestinal mucosal cells, menstruation and other blood losses. Daily iron absorption may increase in response to increased iron demand (eg, growth, pregnancy or blood loss).
Dietary non-haem iron primarily exists in an oxidised (Fe3+) form that is not bioavailable and must first be reduced to the Fe2+ form by a ferrireductase enzyme, which uses vitamin C as coenzyme, before being transported across the intestinal epithelium. This is accomplished by a carrier protein called divalent metal transporter 1 (DMT1), which also traffics other metal ions such as zinc, copper and cobalt by a proton-coupled mechanism.3 ,4 The absorption of non-haem iron can be diminished by co-administration of tretracyclines, proton pump inhibitor and antacid medication, phytates (high-fibre diets), calcium and phenolic compounds (coffee, tea). In addition, Helicobater pylori infection produces gastric atrophy, which can lead to profound iron-deficiency anaemia.5
Haem iron is absorbed into the enterocyte by a putative, not completely identified, haem carrier protein 1.6 Once internalised in the enterocyte, it is likely that most dietary haem is metabolised by haem oxygenase to release Fe2+, which enters a common pathway with dietary non-haem iron before it leaves the enterocyte2 ,3 (figure 1). However, it remains uncertain whether some intact haem traverses the cell, leaving the enterocyte through the action of the recently characterised haem exporters, which are also expressed in kidney, liver and erythroblast, suggesting that they may act at those sites.7 Plasma haem is scavenged and transported by haemopexin to hepatocytes for degradation.
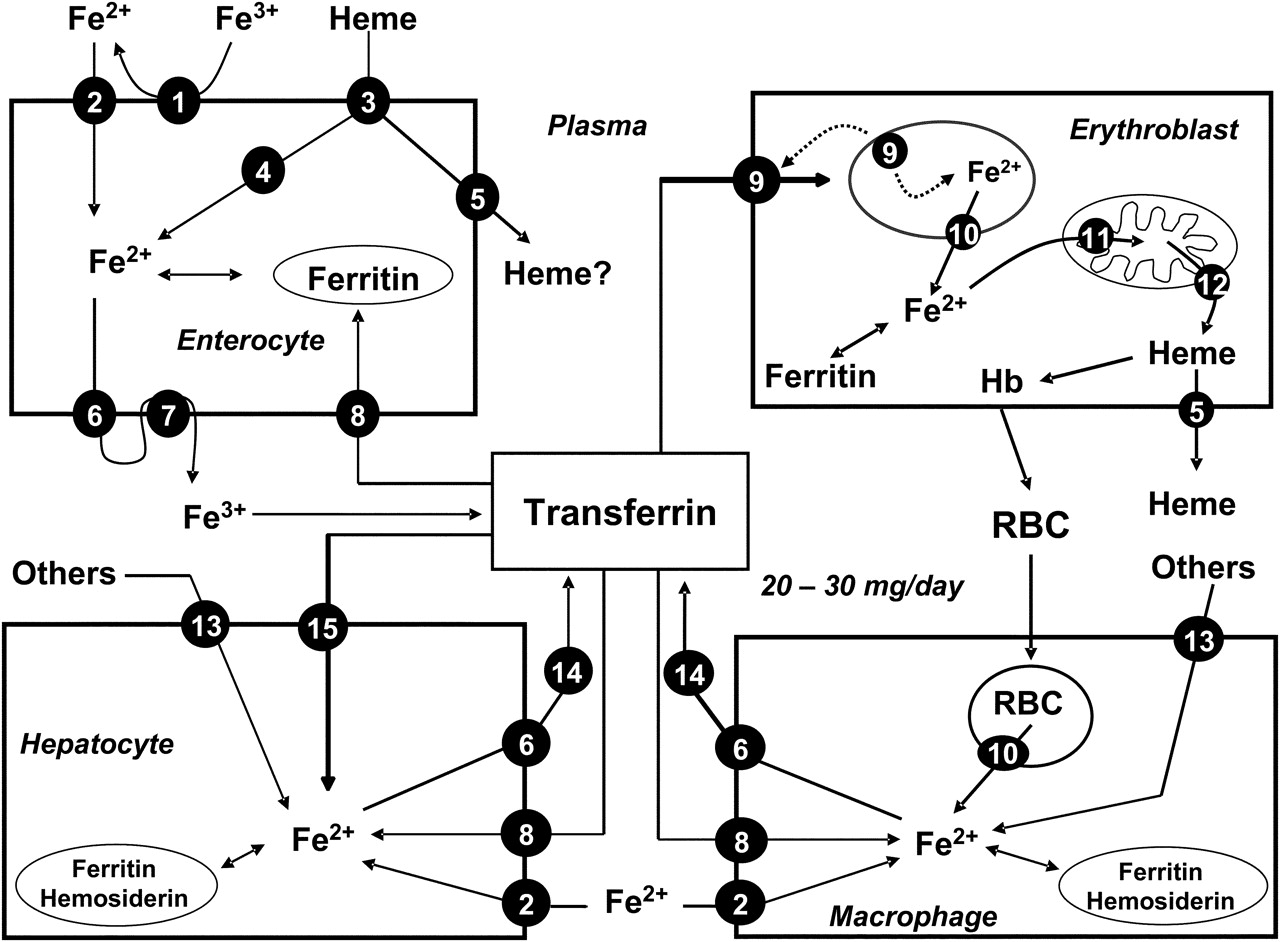
Main pathways of iron metabolism in mammals (modified from Muñoz et al 2). Key: 1, ferrireductase; 2, divalent metal transporter (DMT1); 3, haem protein carrier 1 (HPC1); 4, haem oxygenase; 5, haem exporter; 6, ferroportin (Ireg-1); 7, hephaestin/caeruloplasmin; 8, transferrin receptor-1 (TfR1); 9, diferric transferring–TfR1 complex; 10, natural resistance macrophage protein (Nramp-1); 11, mitoferrin; 12, mitochondrial haem exporter (Abcb6); 13, others: bacteria, lactoferrin, haemoglobin–haptoglobin, haem–haemopexin, etc; 14, caeruloplasmin; 15, transferrin receptor-2 (TfR2). See text for further details.
Once inside the intestinal epithelial cell, most Fe2+ is exported by ferroportin 1 across the basolateral membrane of the enterocyte (absorbed iron), and oxidised to Fe3+ by hephaestin before being bound by plasma transferrin. Ferroportin 1 is also expressed in hepatocytes, reticuloendothelial macrophages and placental syncytiotrophoblasts (where it regulates iron entry into fetal circulation).8
Iron distribution
Iron released into the circulation binds to transferrin and is transported to sites of use and storage. About 30–40% of the iron-binding capacity of transferrin is used in normal physiological conditions; thus transferrin-bound iron is ∼4 mg, but this is the most important dynamic iron pool.9 Transferrin-bound iron enters target cells—mainly erythroid cells, but also immune and hepatic cells—through a highly specific process of receptor-mediated endocytosis (figure 1). As diferric transferrin binds to transferrin receptor 1 (TfR1) at the plasma membrane, patches of cell-surface membrane carrying receptor–ligand complexes invaginate to form clathrin-coated endosomes (siderosomes).9 After clathrin is removed, the siderosomes become acidified through ATP-dependent proton influx, mediated by a family of STEAP (six transmembrane epithelial antigen of the prostate) proteins, leading to conformational changes in both transferrin and TfR1, and promoting the release of Fe3+ from transferrin. Fe3+ is then reduced to Fe2+ by a ferrireductase and transported to the cytoplasm through the DMT1, whereas TfR1 is recycled to the cell membrane and transferrin is shed back into the circulation2 ,10 (figure 1). Within the erythroblast, the mitochondrial iron importer, mitoferrin, plays a critical role in supplying iron to ferrochelatase for insertion into protoporphyrin IX to form haem (the final step of mitochondrial haem synthesis)10 (figure 1). There is some evidence that, in developing erythroid cells, iron could be delivered directly from the siderosomes into the mitochondria.11 Finally, haem exporters transfer haem from the mitochondria to the cytosol and remove excess haem from the erythroid cells (figure 1).
Iron storage
Haemoglobin iron has a substantial turnover, as senescent erythrocytes are phagocytosed by RES macrophages. Within the phagocytic vesicles, haem is metabolised by haem oxygenase, and the released Fe2+ is exported to the cytoplasm through the action of NRAMP1 (natural resistance-associated macrophage protein-1), a transport protein similar to DMT1 (figure 1). Macrophages can also obtain iron from bacteria and apoptotic cells, from plasma through the action of DMT1 and TfR1, and from other sources (figure 1). In the cell, iron can be stored in two forms: in the cytosol as ferritin, and, after breakdown of ferritin, in the lysosomes as haemosiderin. Haemosiderin represents a very small fraction of normal body iron stores, mostly in macrophages, but increases dramatically during iron overload.9 Importantly, iron storage in the macrophages is safe, as it does not lead to oxidative damage.
Iron export from macrophages to transferrin is accomplished primarily by ferroportin 1, the same iron-export protein as expressed in the duodenal enterocyte, and caeruloplasmin2 (figure 1). The amount of iron required for daily production of 300 billion red blood cells (20–30 mg) is provided mostly by macrophage iron recycling.3 Therefore, as daily absorption (1–2 mg) just balances daily loss, internal turnover of iron is essential to meet the bone marrow requirements for erythropoiesis.1–3
The liver is the other main storage organ for iron, and the generation of free radicals and lipid peroxidation products in iron-overload states may result in progressive hepatic tissue injury and eventually cirrhosis or hepatocellular carcinoma.12 The uptake of transferrin-bound iron by the liver from plasma is mediated by TfR1 and TfR2 (figure 1), although it can acquire iron from non-transferrin-bound iron (by a carrier-mediated process consistent with DMT1), ferritin, haemoglobin–haptoglobin complexes and haem–haemopexin complexes. Iron is sequestrated in hepatocytes predominantly in the form of ferritin or haemosiderin. In contrast, once again, ferroportin 1 is likely to be the only protein mediating the export of iron from hepatocytes, which is then oxidised by caeruloplasmin and bound to transferrin2 ,3 (figure 1).
Iron storage in cardiomyocytes is also of great interest, as cardiac failure is the leading cause of death among patients with untreated hereditary haemochromatosis or transfusion-associated iron overload.2 In cardiac cells, excess iron may result in oxidative stress and alteration of myocardial function due to DNA damage by hydrogen peroxide through the Fenton reaction.2
Regulation of iron homoeostasis
The absorption of iron by duodenal enterocytes is dependent on body iron stores, hypoxia, inflammation and rate of erythropoiesis. Two regulatory models have been proposed, both of which may contribute to the regulation of iron absorption: the crypt programming model and the hepcidin model. The crypt programming model proposes that enterocytes in the crypts of the duodenum take up iron from the plasma, via TfR1 and TfR2. The intracellular iron concentration controls the interaction of cytosolic iron regulatory proteins (IRPs) 1 and 2 with iron-responsive elements (IREs). In the absence of iron, IRP1 binds to IREs of TfR1, DMT1 and ferroportin 1 mRNA, the transcript is stabilised, translation proceeds, and the proteins are synthesised. Thus, a high IRP-binding activity reflects low body iron stores and results in upregulation of these proteins in the duodenum increasing dietary iron absorption. When IRPs bind to IREs of ferritin mRNA, translation of the transcript is blocked and synthesis is halted. Thus, ferritin concentrations are reciprocally regulated, being increased in iron-replete states and decreased in iron-deplete states.2
The hepcidin model proposes that hepcidin is produced mainly by hepatocytes in response to the iron content of the blood. Then, hepcidin is secreted into the bloodstream and interacts with villous enterocytes to regulate the rate of iron absorption by controlling the expression of ferroportin 1 at their basolateral membranes. The binding of hepcidin to ferroportin 1 initially causes Janus kinase 2-mediated tyrosine phosphorylation of the cytosolic loop of the carrier protein, phosphorylated ferroportin 1 is then internalised, dephosphorylated, ubiquitinated and ultimately degraded in the late endosome/lysosome compartment. Ferroportin 1 molecules present in macrophages and liver are also targets for hepcidin.13
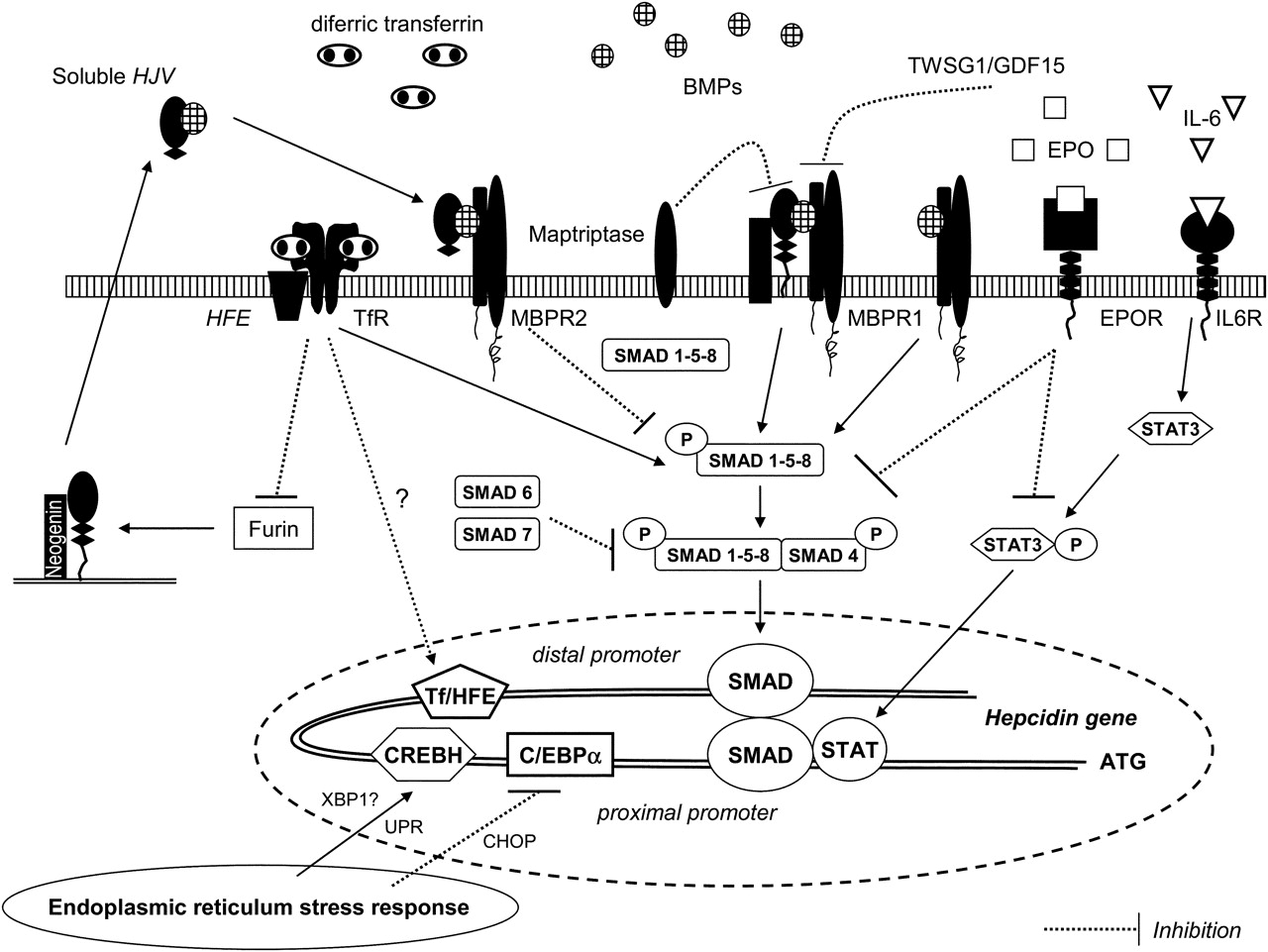
Within normal iron concentration ranges, the sensing process probably involves the local iron-induced production of bone morphogenic proteins (BMPs) such as BMP6. Briefly, BMP6 binds hepatocyte cell surface BMP receptors (BMPRs) I and II together with the BMP coreceptor, haemojuvelin (HJV), initiating a signal that is conveyed intracellularly by phosphorylation of small mothers against decapentaplegic (Smad) proteins. Phosphorylated Smad1, Smad5 and Smad8 form a complex with the common mediator Smad4, before translocation to the nucleus and activating hepcidin expression14 ,15 (figure 2). The soluble form of HJV (sHJV), the release of which (HJV shedding) is inhibited by increasing extracellular concentrations of iron, is believed to compete for BMPR binding with its membrane-anchored counterpart, thereby providing iron-sensitive modulation of hepcidin expression16 (figure 2). Iron may also stimulate the expression of other mediators and modulators, such as Smad6 and Smad7, which seem to attenuate the signal for hepcidin activation (figure 2).

Multiple pathways for hepcidin regulation (adapted from references Pietrangelo19, Lee and Beutler20 and Wessling-Resnick45). BMP, bone morphogenic protein; BMPR, BMP receptor; C/EBPα, CCAAT/enhancer-binding protein; CHOP, CCAAT/enhancer-binding homologous protein (negative regulator of C/EBPα); CREBH, cAMP-responsive element binding protein H; EPO, erythropoietin; EPOR, EPO receptor; GDF15, growth differentiation factor 15; HFE, HFE gene product; HJV, haemojuvelin; IL-6, interleukin-6; IL6R, IL-6 receptor; STAT, signal transducer and activator of transcription; Tf, transferrin; TWSG1, twisted gastrulation; UPR, unfolded protein response; XBP-1, X-box binding protein 1 (endoplasmic reticulum stress-activated transcription factor). See text for further details.
The hepcidin regulatory signalling pathways may also be regulated by HFE. The HFE gene product (together with β2 microglobulin) associates with both TfR1 and TfR2, but it is thought that the HFE–TfR2 complex senses transferrin saturation to modulate hepcidin transcription.17 In this model, diferric transferrin and HFE compete for binding to TfR1. In the presence of high concentrations of diferric transferrin, HFE is thought to dissociate from TfR1 and form part of an iron-sensing complex through its interaction with TfR2.17 ,18 The interaction of transferrin–HFE with TfR2 may signal the high iron content of the blood to an iron sensor and signal-transduction effector complex, possibly the BMP–HJV complex or a distinct system.19 In this regard, binding of diferric transferrin to TfR2 inhibits HJV cleavage by furin, thus inhibiting the release of soluble HJV and resulting in cell-surface HJV-mediated enhanced responsiveness to BMPs and raised hepcidin concentration20 (figure 2). Data from liver tissue of male patients with homozygote HFE mutations and significant iron overload showed appropriate induction of BMP6, but reduced Smad1/Smad5/Smad8 phosphorylation relative to hepatic iron burden and failure of upregulation of hepcidin. Moreover, upregulation of Smad6 and Smad7 occurred in these patients, identifying these molecules as potential aggravators of disease pathogenesis21 (figure 2).
There are also a number of factors that may downregulate liver hepcidin expression. Matriptase-2, a transmembrane serine protease encoded by the gene TMPRSS6, is predominantly expressed in liver, and has a critical role in iron homoeostasis. Matriptase-2 interacts with HJV and inhibits hepcidin synthesis by cleaving membrane HJV into fragments, which presumably abolishes its function22 (figure 2). Homozygous or compound heterozygous mutations in TMPRSS6 leading to hepcidin overexpression have been linked to familial iron-deficiency anaemia refractory to oral iron.23 ,24
It has been known for a long time that elevated erythropoiesis increases iron absorption regardless of body iron loading, and this process seems to be mediated by erythropoietin (EPO), growth differentiation factor 15 (GDF15) and twisted gastrulation (TWSG1).8 EPO suppression of hepcidin expression is mediated through the downregulation of (a) signal transducer and activator of transcription 3 (Stat3) phosphorylation triggered by lipopolysaccharides and (b) Smad1/5/8 phosphorylation induced by iron25 (figure 2). A recent study showed that EPO administration in humans causes a marked and prolonged reduction in circulating hepcidin.26 In addition, there is evidence that EPO upregulates DMT1 and hephaestin gene expression in the duodenum27
GDF15 and TWSTG1 are members of the TGF-β superfamily and are released under conditions of iron-restricted erythropoiesis. GDF15 is secreted by haemoglobinised erythroblasts during the final stages of erythropoiesis, which has been demonstrated to suppress hepcidin expression in vitro,28 whereas TWSTG1, which is produced mainly by the immature erythroid precursors during the early stages of erythropoiesis, suppresses the upregulation of hepcidin expression induced by BMPs29 (figure 2).
Hypoxia-inducible factors (HIFs) are multi-subunit transcription factors that are regulated by hydroxylation of an unstable α subunit controlling its degradation. This modification involves prolyl hydroxylases requiring both oxygen and iron; thus, under hypoxic or low-iron conditions, the prolyl hydroxylase activity is inhibited, resulting in the accumulation and translocation of HIF into the nucleus. HIF binding to the promoter of hepcidin leads to the suppression of hepcidin expression in hepatocytes, whereas its binding to the promoter of DMT1 increases DMT1 expression in enterocytes.30 ,31
In summary, it is hypothesised that, when hepcidin concentrations are increased in iron overload, iron release from intestinal crypt cells (and also from liver and macrophages) is reduced. Hepcidin concentrations are also increased during inflammation, regardless of the patient's iron status (see below). In contrast, when hepcidin concentrations are reduced, as in iron deficiency, anaemia or hypoxia, it is likely that ferroportin 1 expression and iron release from intestinal cells, liver and cells of the RES are increased.8 Genetic abnormalities in HFE, HJV, hepcidin and TfR2 lead to disproportionately low hepcidin concentrations and iron overload (haemochromatosis type I, IIa, IIb and III, respectively). In contrast, mutations in the ferroportin 1 gene are responsible for type IV haemochromatosis.
In erythroid precursors, expression of TfR1, DMT1 and ferritin are reciprocally regulated through IRP1 and IRP2, which act on the IREs present in their RNAs. Thus, when erythropoiesis is stimulated and increased iron supply is needed, expression of TfR1 and DMT1 is increased, whereas synthesis of ferritin is halted.2 Erythroid cells also contain a DMT1 –IRE isoform, which is regulated by miR-Let-7d, a member of a new class of small non-coding single-stranded RNAs of ∼22 nucleotides, the so-called microRNAs. MiR-Let-7d shows an opposite expression trend to DMT1 –IRE. When microRNA concentrations increase, the expression of DMT1 –IRE is inhibited and vice versa. With respect to DMT1 +IRE, the expression of the DMT1 –IRE isoform increases significantly more at the last stage of erythroid differentiation.32 In addition, there is evidence that EPO activates IRP-1, leading to upregulation of TfR1 expression in the erythroid precursors, which is maintained throughout the differentiation process.27 So far, several patients have been reported with DMT1 mutations causing microcytic hypochromic anaemia, due to decreased erythroid iron utilisation, but leading to increased liver iron stores.33
As stated above, senescent red blood cells are degraded by the proteolysis machinery of the phagolysosome in macrophages. The haem molecule reaches the cytosol, where it can act as a sensor molecule regulating the transcription of several genes, or be degraded by haem oxygenase 1 to release its iron. This iron will subsequently be stored in ferritin molecules for further use or exported back to the plasma by ferroportin 1. In macrophages, as well as duodenal enterocytes, ferroportin 1 is mostly found at the plasma membrane, but it is also present in intracellular vesicles after stimulation of its synthesis by intracellular iron.
A previous study carried out on primary cultures of bone marrow-derived macrophages had already proposed that erythrophagocytosis induces ferroportin 1, haem oxygenase 1 and ferritin mRNA expression by a haem-mediated pathway, followed by stimulation of ferroportin 1 and ferritin mRNA translation by an iron-dependent mechanism.34 More recently, Marro et al 35 have shown that ferroportin 1 is transcriptionally coregulated with haem oxygenase 1 by haem. The protoporphyrin ring of haem is sufficient to increase ferroportin 1 gene transcriptional activity, while the iron released from the haem moiety controls ferroportin 1 translation involving the IRE in the 5' untranslated region. These findings suggest that haem controls macrophage iron recycling to ensure the coordinated degradation of haem by haem oxygenase 1, iron storage and detoxification by ferritin, and iron export by ferroportin.
However, the mechanisms controlling the distribution of iron between the ‘storage’ compartment and the ‘exportation’ compartment have not been fully elucidated. It has been proposed that the net amount of ferroportin 1 present at the cell membrane, which is tuned to the erythroid iron needs by systemic signalling, facilitates iron mobilisation from the ferritin-associated iron stores.36 In this regard, EPO has been shown to reduce iron retention in macrophages by decreasing hepcidin and DMT1 expression and increasing ferroportin 1 expression.26 ,37
On the other hand, besides these intrinsic regulations, the amount of ferroportin 1 at the plasma membrane is tightly controlled by systemic regulation relying on ferroportin 1–hepcidin interaction. As for duodenal enterocytes, the binding of hepcidin to ferroportin 1 at the macrophage membrane causes its internalisation and degradation via a Janus kinase 2–clathrin–ubiquitin-mediated pathway. In addition, an independent lipid raft-mediated pathway for ferroportin internalisation and degradation has been recently described as a new route for hepcidin-mediated regulation of ferroportin 1 in macrophages.38 Nevertheless, it has been described that macrophages respond more acutely to a hepcidin challenge than the duodenal enterocytes, and this observation appears to be fully consistent with the central role of macrophages in maintaining body iron homoeostasis.39
As stated above, the liver is another main storage organ for iron. In iron overload, TfR1 is downregulated in hepatocytes.5 TfR2, which is highly expressed in human liver, lacks an IRE and thus is not reciprocally regulated in response to the concentration of plasma iron. Instead, TfR2 protein expression is regulated by transferrin saturation, and is upregulated in iron overload. In normal and iron-loaded conditions, expression of TfR2 exceeds that of TfR1, suggesting that TfR2 plays an important role in hepatic iron loading in haemochromatosis.1
Effects of inflammation on iron homoeostasis and erythropoiesis
Anaemia is a common complication of chronic inflammatory diseases (eg, cancer, rheumatoid arthritis, inflammatory bowel diseases, congestive heart failure), as well as of sepsis and chronic renal failure. This anaemia may be the result of activation of the immune system by the underlying process, and certain immune and inflammatory cytokines including tumour necrosis factor alpha (TNFα), interferon gamma (IFNγ) and interleukins (IL) 1, 6, 8 and 10.40 ,41
As for anaemia of chronic disease (ACD), several pathophysiological mechanisms (cytokines) may be involved41 (figure 3):

{kind=link}
{kind=link}
{kind=link}
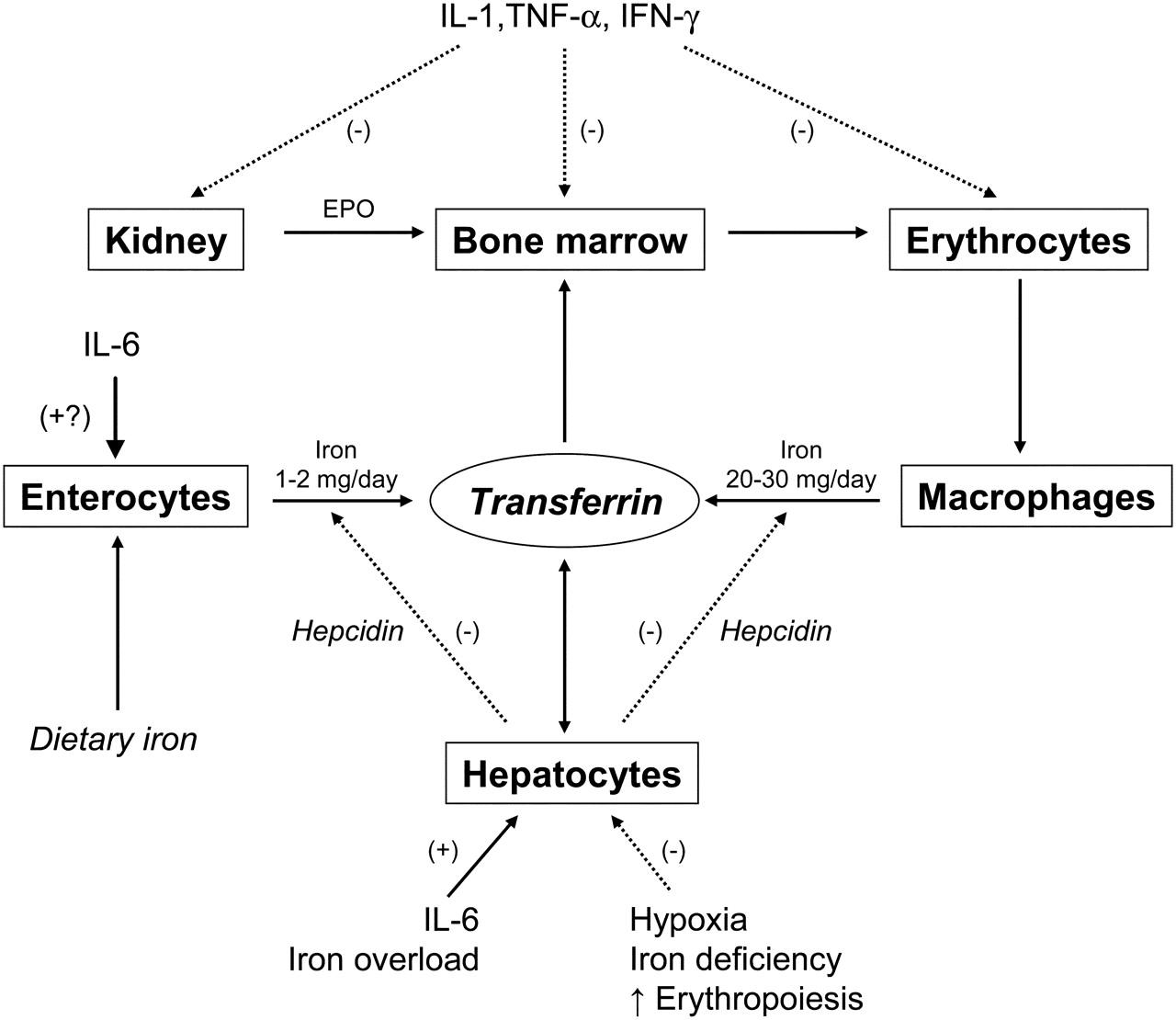
Effects of inflammation on erythropoiesis and iron homoeostasis in mammals (modified from Muñoz et al 2). (–), negative effect; (+), positive effect; EPO; erythropoietin; IFN, interferon; IL, interleukin; TNF, tumour necrosis factor. See text for further details.
-
Decreased red blood cell half-life due to dyserythropoiesis, red blood cell damage, and increased erythrophagocytosis (TNFα).
-
Inadequate EPO responses for the degree of anaemia in most, but not all, patients, such as those with systemic-onset of juvenile chronic arthritis (IL-1 and TNFα).42
-
Impaired responsiveness of erythroid cells to EPO (IFNγ, IL-1, TNFα, hepcidin).43
-
Inhibited proliferation and differentiation of erythroid cells (IFNγ, IL-1, TNFα, and α1-antitrypsin).
-
Pathological iron homoeostasis due to increased DMT1 (IFNγ) and TfR (IL-10) expression in macrophages, reduced ferroportin 1 expression (IFNγ- and IL-6-induced high hepcidin concentrations) in enterocytes (inhibition of iron absorption) and macrophages (inhibition of iron recirculation), and increased ferritin synthesis (TNFα, IL-1, IL-6, IL-10) (increased iron storage)
Inflammatory cytokines such as IL-6 upregulate hepcidin transcription through activation of Janus kinases that phosphorylate Stat3, which become activated.44 Translocation of Stat3 to the nucleus and its binding to the signal transducer and activator of transcription (Stat) responsive element located in the proximal promoter of the hepcidin gene results in increased hepcidin release. This element appears to be regulated through the nearby BMP-responsive element via Smad activation, which is required for full promoter activity. A distal BMP-responsive element site is thought to interact with the Smad–Stat complex, which brings distal and proximal regions of the hepcidin promoter into physical contact.45 Thus, it seems that Smad signalling plays a key role in staging the appropriate response to inflammation. It has been further proposed that Stat3 activation itself, without inflammation, can regulate hepcidin concentrations (eg, patients with glycogen storage disease type 1a who have hepatic adenomas overexpressed hepcidin due to Stat3 activation)46 (figure 2).
Stress pathways signalling through the cellular endoplasmic reticulum unfolded protein response have also been found to induce hepcidin expression. The unfolded protein response has been implicated in the hepatic acute-phase response to lipopolysaccharide, IL-6 and IL-1β, suggesting that hepcidin gene expression may be regulated through an additional layer of endogenous control during inflammation47 ,48 (figure 2). All these lead to low serum iron and decreased transferrin saturation despite normal or high serum ferritin, through iron diversion to the RES (functional iron deficiency, FID), iron-restricted erythropoiesis and mild-to-moderate anaemia.
However, the pathophysiology of acute inflammation-related anaemia (eg, trauma, surgery) is somewhat different. In this setting, inflammatory responses are mediated mainly by IL-6 and IL-8 with transient contribution from TNFα and IL-1 in some visceral surgeries, such as gastrointestinal or cardiac procedures, whereas plasma IFNγ concentrations are undetectable or within the normal range.49–51 Therefore, in most of these conditions, the two major mechanisms leading to anaemia are perioperative or traumatic blood loss and blunted erythropoiesis due to decreased iron availability, whereas EPO concentrations are normal or near normal.52
In the event of persisting decreased iron absorption and/or chronic blood loss, ACD may evolve to anaemia of chronic disease with true iron deficiency (ACD+ID). Interestingly, patients with ACD+ID have significantly lower hepcidin concentrations than those with ACD, and are able to absorb some dietary iron from the gut and to mobilise some iron from macrophages. Thus, circulating hepcidin concentrations in ACD+ID seems to be more responsive to the erythropoietic demands for iron than to inflammation.53
Finally, it must be borne in mind that iron is not only required for erythropoiesis and oxidative metabolism. Cellular immune responses are also dependent on the presence of iron, and specific defects in cell-mediated immunity have been described in detail, even in mild iron deficiency, including the impaired proliferation and function of lymphocytes and natural killer cells, and a depressed neutrophil respiratory burst.54 ,55 For this reason, systemic inflammatory response episodes last longer in critically ill patients with FID, with prolonged stay in the intensive care unit and increased morbidity.56 The effectiveness of the administration of iron sucrose, alone or in combination with recombinant human EPO, was assessed in a population of anaemic critically ill patients.57 Compared with those in the control group who only received folic acid, both treatments resulted in a trend to reduce the requirements for allogeneic blood transfusion (ABT). In addition, patients treated with iron sucrose experienced amelioration of the systemic inflammatory response, as assessed by C-reactive protein concentrations, and a trend to lower mortality than those from the control group. These beneficial effects were not as evident in patients receiving iron sucrose plus recombinant human EPO, as blood marrow stimulation by recombinant human EPO may cause further FID.
Thus, iron deficiency or FID may lead not only to blunted erythropoiesis and chronic fatigue but also to an inappropriate immune response. On the other hand, the evidence that iron overload (or iron supplementation in iron-replete individuals) can promote both infectious and chronic inflammatory diseases is also clear. Thus, iron treatment in patients with active infectious diseases or ongoing bacteraemia should be avoided.
Take-home messages
-
Total body iron is about 3500 mg (50 mg/kg body weight), of which 65% is distributed in red blood cell haemoglobin (2300 mg).
-
Absorption of dietary iron (1–2 mg/day) is tightly regulated, and just balanced against iron loss. Thus, internal turnover of iron is essential to meet the requirements of erythropoiesis (20–30 mg/day).
-
Macrophages and liver are the main iron-storage sites (ferritin), but transferrin-bound iron (3–4 mg) is the most important functional iron pool.
-
Hepcidin, which modulates the expression of ferroportin, is the main iron-regulatory hormone of iron metabolism, and its synthesis is controlled by multiple signalling pathways (eg, inflammation, hypoxia, erythropoietin).
Interactive multiple choice questions
-
This JCP best practice article has an accompanying set of multiple choice questions (MCQs). To access the questions, click on BMJ Learning: Take this module on BMJ Learning from the content box at the top right and bottom left of the online article. For more information please go to: http://jcp.bmj.com/education Please note: the MCQs are hosted on BMJ Learning—the best available learning website for medical professionals from the BMJ Group. If prompted, subscribers must sign into JCP with their journal's username and password. All users must also complete a one-time registration on BMJ Learning and subsequently log in (with a BMJ Learning username and password) on every visit.
References
Footnotes
-
All author contributed equally to the design, writing and discussion of this paper.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.