Article Text

Abstract
Pulmonary fibrosis in surgical lung biopsies is said to have a ‘usual interstitial pneumonia-pattern’ (UIP-pattern) of disease when scarring of the parenchyma is present in a patchy, ‘temporally heterogeneous’ distribution. These biopsies are one of the more common non-neoplastic specimens surgical pathologists encounter and often pose a number of challenges. UIP is the expected histopathological pattern in patients with clinical idiopathic pulmonary fibrosis (IPF), but the UIP-pattern can be seen in other conditions on occasion. Most important among these are the rheumatic interstitial lung diseases (RILD) and chronic hypersensitivity pneumonitis (CHrHP). Because theses entities have different mechanisms of injury, approach to therapy, and expected clinical progression, it is imperative for the surgical pathologist to correctly classify them. Taken in isolation, the UIP-pattern seen in patients with IPF may appear to overlap with that of RILD and CHrHP, at least when using the broadest definition of this term (patchy fibrosis). However, important distinguishing features are nearly always present in our experience, and the addition of a multidisciplinary approach will often resolve the critical differences between these diseases. In this manuscript, we review the distinguishing clinical, radiologic and histopathological features of UIP of IPF, RILD and CHrHP, based, in part, on the existing literature, but also lessons learned from a busy lung biopsy consultation practice.
- RHEUMATOLOGICAL PATHOLOGY
- PULMONARY PATHOLOGY
- RHEUMATOID ARTHRITIS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The diffuse parenchymal lung diseases (commonly referred to as ‘interstitial lung diseases’ or ‘ILDs’) include a heterogeneous group of mainly non-neoplastic disorders. ILDs are mostly inflammatory in nature and the resulting injury to the lung parenchyma can lead to fibrosis and eventual honeycomb cystic remodelling. Some ILDs are characterised by the presence of fibrosis. Dominant among these is ‘usual interstitial pneumonia’ (UIP) which is the most common histopathological form of diffuse lung fibrosis occurring in older adults, first described by Liebow in 1969.1 According to Liebow, UIP was idiopathic in about half the affected patients, and the idiopathic form of UIP was referred to clinically as ‘idiopathic pulmonary fibrosis’ (IPF). The other half of UIP patients had other causes of diffuse lung fibrosis. The common use of the term UIP by pathologists to describe diffuse fibrosis is often confusing to clinicians who tend to assume that a pathological designation of UIP denotes a diagnosis of IPF, not realising that some forms of the UIP-pattern of advanced fibrosis can have other aetiologies. Our goal in this review is to shed light on the multidisciplinary clues that are most helpful in distinguishing those forms of UIP that are manifestations of pulmonary fibrosis from other causes.
From a histopathological perspective, the UIP-pattern of fibrosis is characterised by two key features:
-
Spatial or geographic heterogeneity which refers to a patchy distribution of dense parenchymal scar alternating with areas of less affected or normal parenchyma (figure 1A).
-
Temporal heterogeneity which refers to areas of densely collagenised fibrosis (the past) with variable smooth muscle proliferation (‘old’ fibrosis) alternating with active fibroblast foci (‘new’ fibrosis; the present). Importantly, the UIP-pattern biopsy must also have the ‘future’, with normal lung evident (figure 1B).

Essential characteristics of the usual interstitial pneumonia-pattern of fibrosis. The geographic (or spatial) heterogeneity is seen at scanning magnification (A, 20×, H&E). Large conglomerate zones of pulmonary fibrosis (at the pleural surface and lower right) alternate with areas of uninvolved parenchyma (lower left). At higher magnification (B, 200×, H&E), the temporal heterogeneity of this active fibrosing process is appreciated by identifying immature fibrosis in the form of fibroblast foci (arrow).
These features provide the minimal diagnostic criteria for inclusion into the UIP-pattern of disease. However, histopathological clues to the aetiology of pulmonary fibrosis reside first in recognising the pattern and distribution of chronic parenchymal damage, and then determining the cellular elements involved in the process. For example, fibrotic parenchymal diseases with upper lung zone predominance implicate an inhalational injury, as does airway-centred scarring seen under the microscope, while fibrosis that begins at the periphery of the lung bases tends to occur in diseases thought to have a systemic immunologic mechanism of injury such as rheumatic disease (RD). An upper lobe distribution of hyaline fibrosis accompanied by small non-necrotising granulomas along lung lymphatic routes is characteristic of sarcoidosis, whereas an upper and mid-lobe inflammatory disease with tiny interstitial granulomas centred on the airways, defines subacute hypersensitivity pneumonitis (HP). Unfortunately, overlaps occur, so the only consistent and reliable approach to these diseases when fibrosis is the dominant element requires a clinical, radiological and histopathological approach.
The three most common diseases that present with advanced pulmonary fibrosis having a UIP-pattern are IPF, rheumatic ILD (RILD), and chronic hypersensitivity pneumonitis (CHrHP). The key clinical, radiographic and histopathological differences that help distinguish these three manifestations of pulmonary fibrosis are presented.
Usual interstitial pneumonia of IPF
Definition
IPF is a specific form of chronic, progressive fibrosis, confined to the lungs and of unknown cause.2 It occurs primarily in older male patients, and is associated with the histopathological and radiological patterns of ‘UIP’.2 ,3 The definition of IPF requires exclusion of other forms of interstitial pneumonia as well as ILDs associated with environmental exposure, drugs or systemic disease.2 ,3 The distinguishing clinical, radiological and histopathological features are discussed below and highlighted in table 1.
UIP in IPF
Distinguishing clinical features
The presenting symptoms are dominated by gradual onset of exertional dyspnoea, sometimes associated with chronic cough. IPF is associated commonly with a history of smoking.4 The chief physical examination characteristic at auscultation is dry crackles at the lung bases (so-called ‘Velcro rales’). Clubbing of the fingers occurs in approximately 40–75% of patients, and it appears to occur in IPF more commonly than in other ILDs.5 There should be no chronic occupational or environmental exposure, clinical evidence of connective tissue disease, such as rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE), or history of drug exposure known to cause ILD (nitrofurantoin, methotrexate, cyclophosphamide, or any of a variety of chemotherapeutic drugs).2
Distinguishing radiological features
The typical radiological pattern of UIP of IPF is heterogenous fibrosis in spatial (normal lung interspersed among areas of fibrosis) and temporal distribution (fibrosis in different stages of evolution). The diagnostic radiological pattern of UIP requires honeycombing with a peripheral predominant, lower lobe, non-segmental distribution (apico-basilar gradient) (figure 2A).6 The upper lobes are involved to a lesser degree, and traction bronchiectasis may be present. The classic pattern of UIP of IPF on CT is diagnostic and biopsy is not warranted if it fulfils these imaging patterns.2

Radiological characteristics of usual interstitial pneumonia in idiopathic pulmonary fibrosis. Sagittal CT sections (A) show characteristic heterogeneous fibrosis and honeycombing in lung bases (arrow heads). Coronal maximum intensity projection (B) shows the typical apico-basilar gradient of fibrosis.
UIP of IPF may also be suggested without CT honeycombing when heterogenous fibrosis is identified in an appropriate clinical setting; lobular distortion is worst in lower lobes, and upper lobe non-segmental involvement is present (figure 2B).7 ‘Microscopic honeycombing’ may be seen in biopsies from these latter patients, presumably as an early manifestation of the macroscopic cysts seen on imaging. Ground-glass abnormalities, if present, are typically limited in extent and restricted to basilar subpleural areas of fibrosis, unless patients present during an acute flare of the disease (so-called ‘acute exacerbation of IPF’).8
Distinguishing histopathological features
In UIP of IPF, the distribution of fibrosis tends to be subpleural and paraseptal (figure 3A). Scattered foci of immature fibrosis (so-called ‘fibroblast foci’) are found at the interface between fibrotic and normal parenchyma (figure 3B) as a ‘bulge’ from the interstitium. These foci must be distinguished from fibroblastic alveolar plugs of organising pneumonia. A true fibroblastic focus should be interstitial-based and never a formed polypoid intrusion into the alveolar space. These active ‘new’ areas of fibrosis coexist with the dense ‘old’ collagen deposition and provide the basis of ‘temporal heterogeneity’ in UIP. Smooth muscle is commonly prominent within areas of advanced confluent fibrosis (figure 3C), especially in subpleural scar. Areas of microscopic honeycomb remodelling (MHR) are present in the vast majority of cases (figure 3D), even in the absence of diagnostic honeycomb cysts on high-resolution CT imaging. These areas likely represent completely destroyed lung lobules devoid of alveoli and characterised by aggregates of small cystic spaces containing mucous. These microcysts are consistently lined by ciliated respiratory epithelium attesting to their terminal airway derivation. Fibrosis surrounds MHR at least on three sides, a helpful feature in distinguishing MHR from more common chronic small airway injury lesions of ‘peribronchiolar metaplasia’ (PBM) where respiratory epithelium extends along alveolar ducts in a limited fashion without well-formed cysts. A small amount of chronic inflammation may attend these, but is not expected away from honeycomb remodelling in fibrosis or in any preserved alveolar walls.

Characteristics of advanced fibrosis in usual interstitial pneumonia of idiopathic pulmonary fibrosis include a subpleural distribution of fibrosis (A, 20×, H&E), relative frequency of fibroblast foci (arrows) and relative absence of any significant inflammatory cell infiltrate (B, 400×, H&E), smooth muscle proliferation in the subpleural scars (asterisk) (C, 40×, H&E), and the frequent occurrence of microscopic honeycomb remodelling (D, 20×, H&E).
UIP-pattern in rheumatic diseases
Definition
All RDs have the potential to produce diffuse lung fibrosis.9 Autoimmune disorders are estimated to be responsible for approximately 25% of ILD deaths and 2% of deaths from all respiratory causes.10 ,11 The most frequent RDs associated with pleural and pulmonary manifestation are: RA, SLE, progressive systemic sclerosis, polymyositis/dermatomyositis and Sjögren syndrome. RA is the most common form of RD worldwide, and by sheer numbers is second only to scleroderma in producing lung disease. However, RA is most frequently implicated in producing a UIP-pattern in lung biopsies.9–12 Readers interested in exploring the other RDs with pleuro-pulmonary manifestations are referred to several excellent reviews.9 ,10 ,13 The distinguishing clinical, radiological and histopathological features are discussed below and summarised in table 2.
UIP in rheumatic disease
Distinguishing clinical features
Advanced pulmonary fibrosis in RD is often apparent in cases in which the connective tissue disease is well established. However, there are also cases in which the presence of ILD may precede the systemic or serological manifestations of the disease by months or even years.10 Interestingly, ILD is more commonly seen in men with RA, despite RA being more common in women.14 High titres of rheumatoid factor and a smoking history are also highly associated with ILD in RA.15 ,16 From a clinical perspective, younger age at onset, unexplained joint or cutaneous manifestations and suggestive serologic abnormalities all play important roles in distinguishing UIP in IPF from UIP-pattern disease in RILD.
Distinguishing radiological features
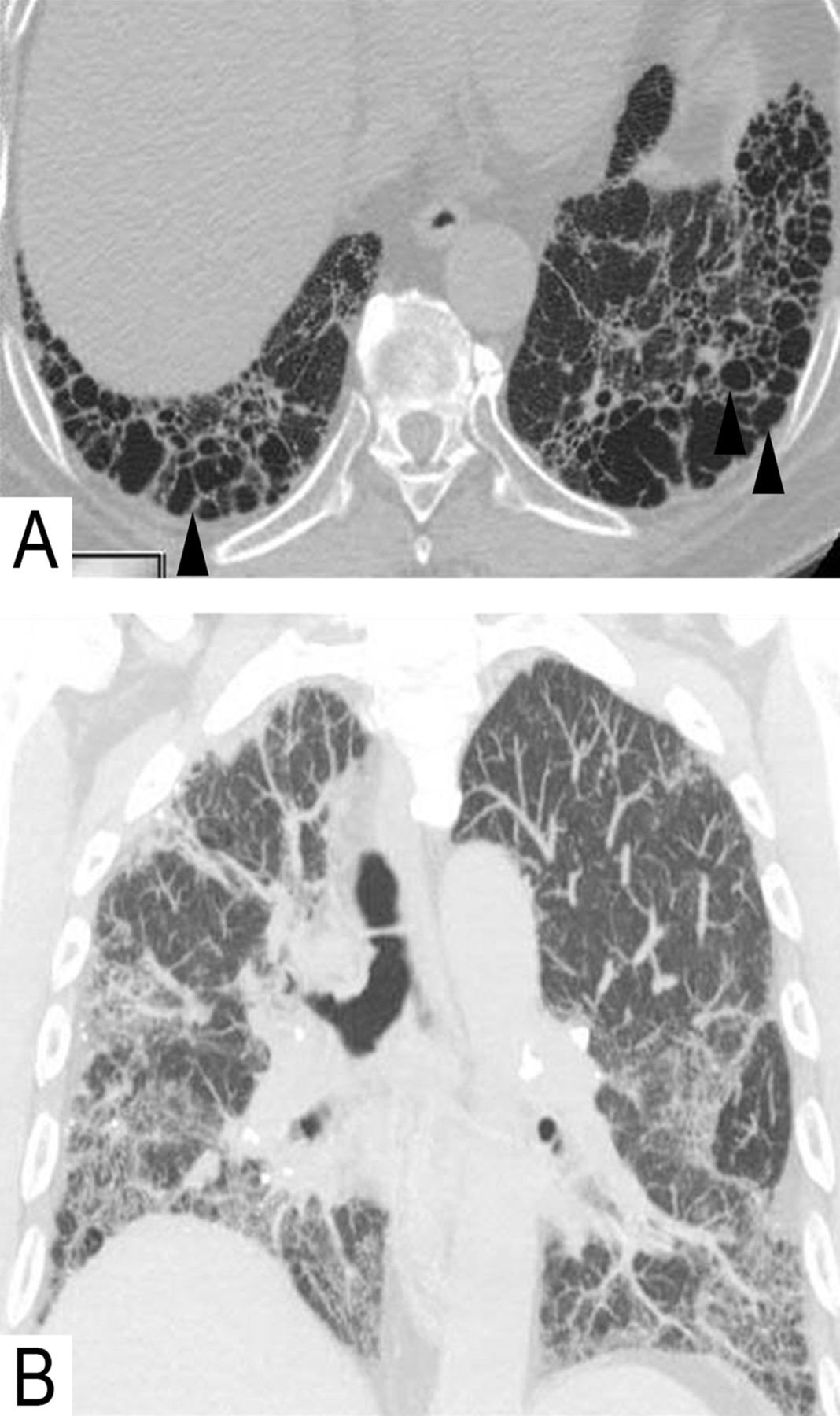
General clues to the diagnosis of UIP in RD are the presence of other radiological clues to RD, including pleural or pericardial effusions (SLE, RA), dilated oesophagus (scleroderma), follicular bronchiolitis/lymphoid interstitial pneumonia (RA, Sjogren), or rarely rheumatoid nodules (RA).17 Interstitial fibrosis can be in the pattern of non-specific interstitial pneumonia (NSIP) or UIP, depending on the underlying disorder, but when a UIP-pattern is present, the prognosis appears to be better than that of UIP in IPF. The common findings are reticular opacities, lobular distortion and traction bronchiectasis or bronchiolectasis (figure 4A). When honeycomb cysts occur in RA, the disease may mimic IPF perfectly, especially if there is no evidence of airways abnormalities, such as fuzzy centrilobular nodules, ground-glass opacities (figure 4B), or bronchial and bronchiolar wall thickening with heterogeneous lung attenuation.

Radiological characteristics of rheumatic interstitial lung disease. Asymmetrical fibrosis (A) with honeycombing (usual interstitial pneumonia) in lung bases (arrows) in a patient with rheumatoid arthritis. In the same patient, the upper lobes (B) showed ill defined centrilobular ground-glass opacities remote from fibrosis, shown to be cellular interstitial inflammatory infiltration on histopathology (arrow head).
Distinguishing histopathological features
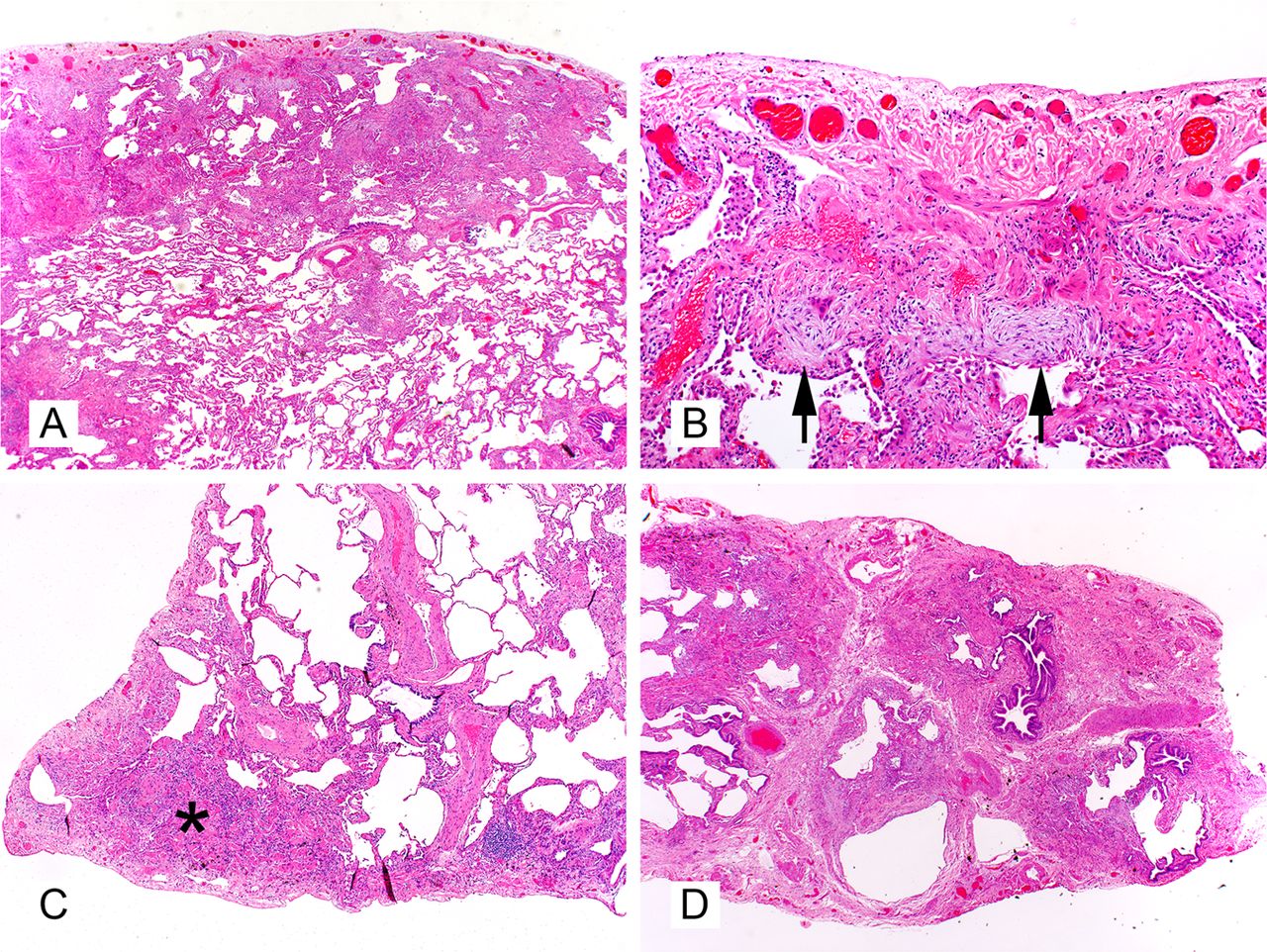
Lymphoid hyperplasia at very low magnification (figure 5A) is a very helpful feature suggesting UIP in RILD as the correct diagnosis. The number of lymphoid follicles and interstitial B and T cells tend to be greater in RA-related UIP than in the idiopathic UIP of IPF.18 ,19 In less fibrotic areas, the lymphoid hyperplasia may be seen in a peribronchiolar distribution with associated PBM (figure 5B). In RILD, less advanced areas of parenchymal fibrosis may show subtle interstitial thickening with either inflammatory cells or fibrosis. This pattern of interstitial involvement suggests a NSIP pattern of injury as the primary event, and strongly argues against UIP of IPF (figure 5C). Surgical pathologists who do not routinely interpret pulmonary wedge biopsies for ILD may mistake these less involved areas for ‘normal lung’, incorrectly supporting a diagnosis of UIP of IPF. Two-thirds of cases of RA show a diffuse fibrosing lung disease with almost equal proportions of UIP and NSIP pattern.20–22 Lymphoid aggregates and follicles with germinal centres follow the airways with resulting chronic follicular bronchiolitis (figure 5D). Another helpful differential characteristic is the presence of overlapping acute, subacute (organising pneumonia) and chronic changes in the same biopsy sample when RILD produce pulmonary fibrosis. Some studies have shown that patients with RILD have fewer fibroblastic foci, smaller honeycomb cysts and less emphysema than patients with IPF.19 Pleural fibrosis is uncommon in IPF (the disease begins in subpleural lung, not pleura), but is typically present in RA and other RDs with thoracic manifestations.

Characteristics of advanced fibrosis in rheumatic diseases include marked lymphoid hyperplasia (A, 20×, H&E), peribronchiolar lymphoid hyperplasia with associated peribronchiolar metaplasia (B, 20×, H&E), presence of an non-specific interstitial pneumonia pattern of disease in the background which may be mistaken as ‘normal’ in comparison with the large zones of fibrosis (C, 20×, H&E), and follicular bronchiolitis (D, 400×, H&E).
UIP-pattern in chronic hypersensitivity pneumonitis
Definition
HP is a diffuse parenchymal lung disease resulting from inhalation of mainly organic antigens by susceptible individuals. Clinical presentations range broadly, based on dose of antigen and frequency/chronicity of exposure, producing acute, subacute, or chronic disease at presentation.23–25 Acute and subacute HP are not relevant to our topic as neither produces a UIP-pattern, and these can be explored elsewhere.26–30 The distinguishing clinical, radiological and histopathological features of CHrHP are discussed below and summarised in table 3.
UIP in CHrHP
Distinguishing clinical features
The clinical history often reveals a temporal relationship between symptoms and some environmental exposure. Potential offending environmental agents may include certain pets, especially birds, exposure to farming-related dusts or fumes, humidifiers, vaporisers, hot tubs, indoor swimming pools and residual water damage to drywall, carpet, or other indoor furnishings.31 The exposure may be occupational or occurring at home. The clinician must take an exhaustive history of work, home and pet exposures, and focus on any exposure to dust, smoke, or fumes of any type. Almost any indoor environment may be a potential source of an antigen causing HP. ChrHP typically presents with an insidious or gradual onset of dyspnoea, cough, fatigue, malaise and chest tightness.31 Physical findings include bilateral basilar crackles or wheezes. Clubbing is uncommon at presentation, but if present, predicts clinical deterioration.30 Serum precipitating antibodies to a variety of commonly implicated antigens have characteristic low sensitivity and specificity. They are helpful when positive, but not helpful when negative. Pulmonary function tests typically show a restrictive pattern with low lung volumes and diffusing capacity.32
Distinguishing radiological features
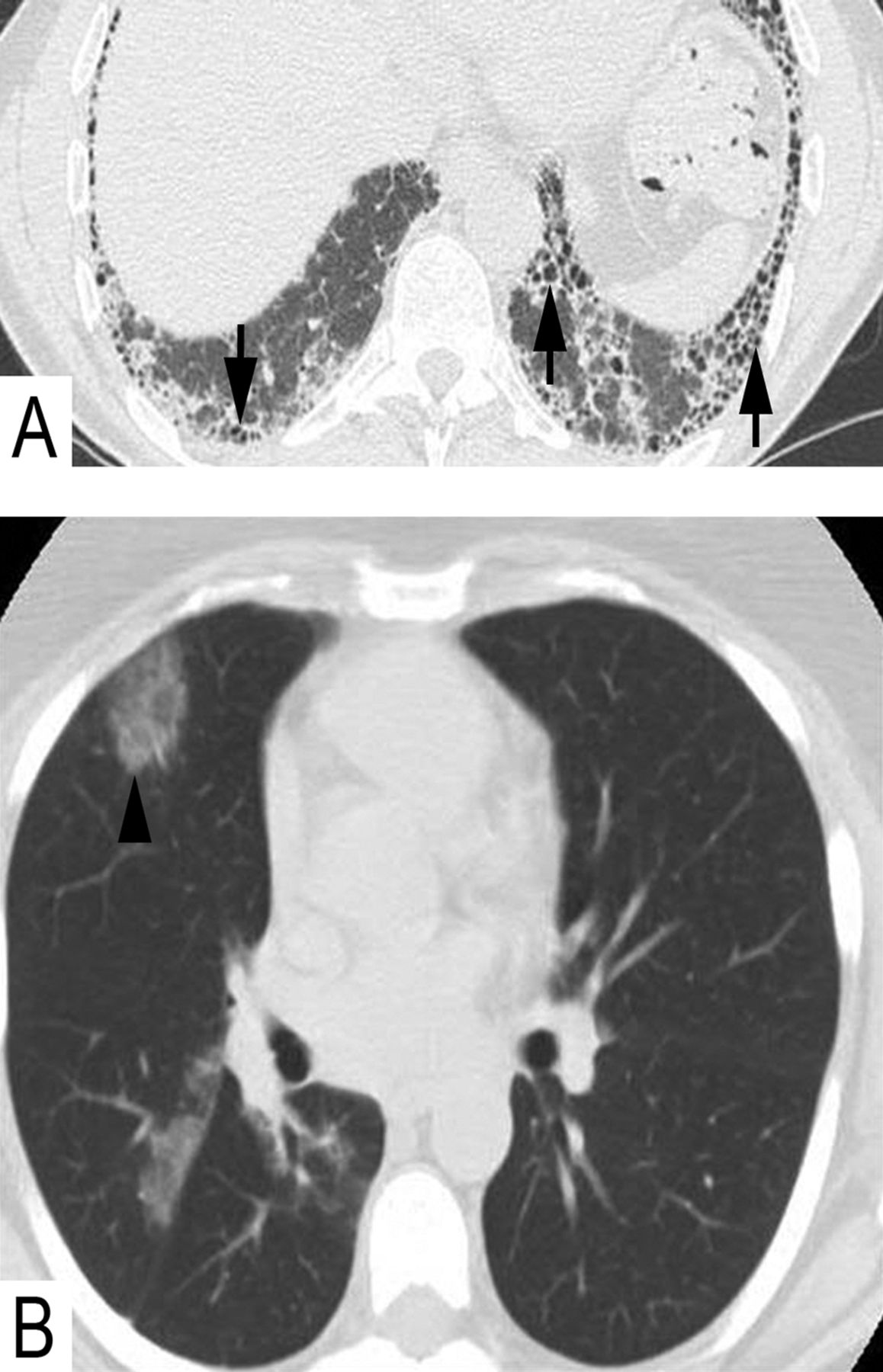
The imaging features of chronic HP are dominated by fibrosis (figure 6A), honeycomb change that involves the lower lobes, and in 80% of cases involvement of the upper lobes simultaneously (figure 6B). Sometimes, honeycomb change is predominant in upper lobes, a finding that when present, is distinctive of UIP in CHrHP.17 Although interstitial fibrosis can have an NSIP or UIP pattern, one of the key distinguishing features in HP is the presence of air-trapping (figure 6B) that is exaggerated on expiration scans due to underlying constrictive bronchiolitis (also referred to as ‘mosaic perfusion’ or ‘mosaic attenuation’).33 ,34

Radiological characteristics of chronic hypersensitivity pneumonitis. There is often fibrosis and honeycombing (A) in the lung bases (arrows) similar to idiopathic pulmonary fibrosis. However, in the same patient (B), there is also fibrosis in the upper lobes (arrows) and air-trapping (asterisk).
Distinguishing histopathological features
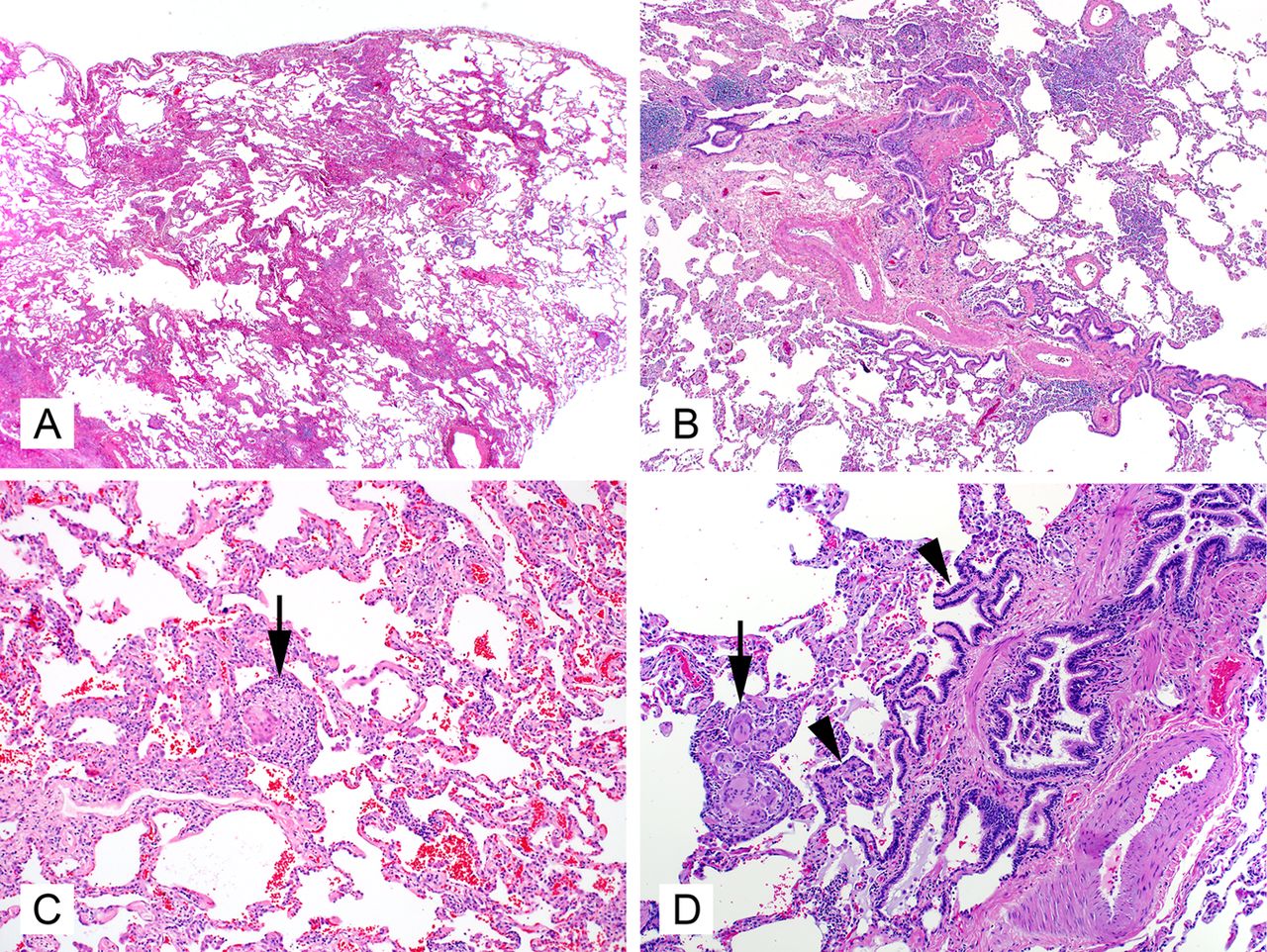
Occasional cases of CHrHP will show advanced fibrosis that is indistinguishable from that seen in UIP of IPF. However, there are several histopathological clues to help suggest CHrHP as the aetiology. Because chronic HP is a result of repeated inhalational exposure, the primary site of injury is the airways. It makes aetiologic sense that most cases of CHrHP will show a centrilobular dominant pattern of fibrosis35 ,36 (figure 7A), as opposed to the subpleural and periseptal fibrosis of UIP of IPF (where centrilobular sparing is the norm). Centrilobular fibrosis is characterised by peribronchiolar fibrosis with bronchiolar distortion and occlusion by scarring and smooth muscle hyperplasia. Respiratory bronchioles are frequently involved. Bridging fibrosis is frequently identified in HP.25 ,37–39 It is represented as linear fibrotic connection between centrilobular and perilobular areas (subpleural or/and paraseptal) or between centrilobular and adjacent centrilobular area. Similarly, the fibroblast foci of CHrHP are typically present at the edge of the fibrosis in peribronchiolar areas.39 ,25 Again, like most inhalational disease, ChrHP affects the upper lobes early in the course of disease. Surgical biopsy from more than one lobe is essential to distinguish UIP of IPF from UIP-pattern in CHrHP.40 PBM is a very common finding in CHrHP (figure 7B), and if it is seen diffusely throughout the biopsy, a careful search for other histopatholological support for CHrHP is warranted.41 ,42 The ‘smoking gun’ of CHrHP is the poorly formed interstitial granuloma, or rare aggregate of interstitial giant cells (figure 7C,D). Unfortunately, the frequency of this finding seems to decrease with increasing fibrosis. Other findings in CHrHP that help to differentiate it from UIP of IPF are the presence of inflammatory bronchiolitis with lymphoplasmacytic infiltration.

Characteristics of advanced fibrosis in chronic hypersensitivity pneumonitis include centrilobular fibrosis with relative sparing of the septum and pleura (A, 20×, H&E), centrilobular interstitial inflammation with extensive peribronchiolar metaplasia (arrow heads) (B and D, 40×, H&E), and rare interstitial giant cells/histiocyte aggregates of poorly formed granulomas (arrows) (C and D, 200×, H&E).
Summary and conclusions
UIP-pattern fibrosis in IPF can often be distinguished from other common forms of diffuse lung fibrosis by close examination of the clinical context, radiological features and histopathology in the individual patient. Figure 8 illustrates the scanning magnification differences between UIP of IPF, RILD and CHrHP. Because several of the key distinguishing features are identifiable at scanning magnification, an attempt to review the whole case initially at low magnification is essential. This should be followed by higher magnification investigation for other findings. Table 4 highlights the key clinical, radiological and pathological features of the most common causes of a UIP-pattern.
Comparison of most common features in practice (authors experience)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Low power examples of usual interstitial pneumonia (UIP) of idiopathic pulmonary fibrosis (IPF), rheumatic interstitial lung disease (RILD), and chronic hypersensitivity pneumonitis (ChrHP). UIP of IPF shows a pauci-cellular subpleural fibrosis with multiple fibroblast foci (A). RILD shows lymphoid hyperplasia with chronic bronchiolitis (B). CHrHP shows bridging centrilobular fibrosis with a mild inflammatory cell infiltrate and peribronchiolar metaplasia (C).
In our experience with a busy ILD consultative practice, common pitfalls include diagnosing UIP of IPF in young patients, not giving enough attention to the robust lymphoid infiltrate of RILD, ignoring the background NSIP-pattern of RILD and CHrHP, and focusing too much on areas of MHR which are really just areas of ‘end-stage’ lung fibrosis, to coin a term commonly used in the renal pathology literature. Identifying the correct diagnosis in this setting has significant implications for therapy today and new therapies tomorrow, even if pulmonary fibrosis from any cause carries significant morbidity and mortality.
Take-home messages
-
Not all cases showing a usual interstitial pneumonia-pattern (UIP-pattern) of fibrosis are secondary to idiopathic pulmonary fibrosis (IPF).
-
A variety of histologic clues often allows a pathologist to discern between IPF and the two other common diseases which show a UIP-pattern of fibrosis, rheumatic interstitial lung disease and chronic hypersensitivity pneumonitis. Clinical and radiological differences, highlighted in this manuscript, can also be used to support pathologic hypotheses.
References
Footnotes
-
Contributors MS was responsible for planning, writing and critically reviewing the work. MD was responsible for planning, writing and critically reviewing the work. PP was responsible for writing and critically reviewing the radiology portions of the work. JP was responsible for writing and critically reviewing the clinical portions of the work. KL was responsible for planning and critically reviewing the work. He is the guarantor.
-
Competing interests None.
-
Ethics approval Approval from the institutional review board was not required in this manuscript as there were no study subjects.
-
Provenance and peer review Commissioned; internally peer reviewed.