Article Text

Abstract
Metastatic colorectal cancer harbouring a mutation in codon 12 or 13 of the KRAS gene does not benefit from therapy with antibodies targeting the epidermal growth factor receptor (EGFR). The implementation of community KRAS testing is generating a rapid flow of new data that have implications for the pathologist and testing guidelines besides the physician. Therefore, it seems timely to draw together the threads of this large body of information in order that pathologists can be knowledgeable partners in the multidisciplinary process of targeted cancer therapy and to help refine current testing guidelines. This review addresses (1) the most relevant methodological and technical aspects of KRAS testing in terms of sample site (primary/metastatic), test specimens (resection/biopsy/cytology) and the diverse molecular methods available; (2) the issues related to daily practice, namely, the timing of the test, its turnaround time and the quality control procedures; and (3) the evidence related to the relationship between KRAS genetic intratumoural heterogeneity, clinical sensitivity of mutational detection tools and anti-EGFR treatment outcome. Hopefully, in the near future, elucidation of the potential of biomarker panels and of the mechanisms underlying primary and acquired resistance to anti-EGFR therapy will refine even further personalised treatment regimens for patients with metastatic colorectal cancer.
- MOLECULAR BIOLOGY
- EGFR
- COLORECTAL CANCER
Statistics from Altmetric.com
Introduction
The epidermal growth receptor (EGFR) is a major therapeutic target in metastatic colorectal cancer (mCRC).1 Cetuximab, a human–mouse chimeric monoclonal antibody (subtype IgG1), and panitumumab, a fully human monoclonal antibody (subtype IgG2κ), are directed against the EGFR and can be used as monotherapy or combined with chemotherapy.1 Retrospective subset analyses of the data from phase II and III clinical trials strongly suggest that patients whose tumour has KRAS mutations in codon 12 or 13 do not benefit from these drugs irrespective of whether they are used as monotherapy2 ,3 or in combination with chemotherapy.4–7 KRAS gene testing is mandatory in mCRC patients in the USA, Europe and Japan,8 and the use of cetuximab and panitumumab is restricted to codon 12 and 13 wild-type tumours. However, an optimal KRAS testing procedure has yet to be established.9 ,10 In fact, procedures vary across laboratories, and the routine approach to KRAS testing differs between Europe (most tests are performed in centralised laboratories) and the USA (in-house testing in most institutions).
Since the introduction of community KRAS testing and quality control programmes in 2008,10 ,11 a large body of data has accumulated on the various facets of this complex topic. Hence, it seems the time is now ripe to review the technical, clinical and therapeutic aspects of KRAS mutation testing also to enable pathologists to be knowledgeable partners in targeted cancer therapy. In addition, a review of the most recent data related to sample selection and processing, the analytical and clinical sensitivity of testing methods and quality control programmes will also help to improve test guidelines.
The sample
Primary and metastatic sites
KRAS mutation status assessment is generally requested by the oncologist upon diagnosis of mCRC. According to current guidelines,9 ,10 biopsy of the metastatic site is not necessary because the test can be reliably performed on the archival tissue blocks containing the surgical resection specimen of the primary tumour. However, when metastatic tissue is available, testing can be performed on either sample. It is well established that KRAS mutation is an early event in colorectal tumours and is highly stable during the course of the disease. Indeed, many studies investigated whether testing the primary tumour predicts the mutation status of the corresponding metastasis, and the concordance was reported to be either almost complete 12–27 or complete.28–33 A recent meta-analysis that included 986 paired primary and distant metastases confirmed these findings.34 A slight difference in concordance has been reported depending on the site of metastasis. In fact, liver metastases nearly always (96.4%) share the KRAS mutational status of the primary tumours, as shown by Knijn et al35 in 305 paired samples. Primary and lymph node metastases34 and primary and lung metastases36 are less often concordant. The discordance in the latter study was 32.4%, which is clinically relevant. Consequently, this should be taken into account in case of isolated lung metastasis from colorectal cancer.
Resection samples and endoscopic biopsies
Diagnostic biopsy specimens represent a tiny fragment of the primary colorectal tumour, which raises the question: ‘Are they sufficiently representative of the tumour to be used to make treatment decisions?’. The same KRAS point mutation was identified in the biopsy and in the corresponding resection specimens in 12/12 cases.37 In a larger series (n=125) of paired samples, concordance of KRAS mutational status between biopsy and resection specimens was very high regardless of the method used.38 More recently, a high concordance between biopsies and resection specimens was reported also for other early driver mutations such as BRAF, PIK3CA and TP53.39 Accordingly, genome-wide sequencing showed that somatic mutations of ‘early’ driver genes are present in all colorectal tumour cells.40
Neoadjuvant chemoradiation therapy, commonly used in rectal cancers, leads to complete tumour regression in approximately 10%–20% of patients and to an almost complete tumour regression in a further 20%–30% of cases.41 Although chemotherapy or radiotherapy do not alter the genetic status of cancer cells,42 KRAS genotyping on post-treatment samples is challenged by the paucity of neoplastic cells.43 In these circumstances, highly sensitive assays coupled with laser capture microdissection that selects a pure population of tumour cells while avoiding contamination from surrounding tissue reduce the possibility of missing a mutation in the KRAS gene.
Cytological samples
In patients with mCRC whose primary tissue is not available or is inadequate, KRAS testing can be reliably performed on cytological specimens taken from metastatic sites.30 To evaluate the suitability of cytology for KRAS testing, we and others have performed validation studies on paired cytological and histological samples. We found a 92.3% concordance rate,44 which is similar to the results reported by Pang et al (87.5%)45 and Bozzetti et al (88%).46 Rapid on-site evaluation of the harvested material by a cytopathologist increases the sample adequacy rates for KRAS testing.34 Cytological samples may also be obtained by touch imprinting fresh tumour tissue against glass slides47 or by applying Whatman Flinders Technology Associates (FTA) cards48—a procedure that results in tumour-rich samples and shortens the KRAS assay turnaround time.49
The method
The KRAS test is performed in the pathologist's laboratory or in a referral centre. In both cases, the pathologist selects the sample and the tissue area to test with the aim of obtaining a percentage of tumour cells acceptable for the assay being used. The College of American Pathologists (CAP)9 and the European Society of Pathology (ESP)10 have recommended standardised morphologic sample assessment prior to DNA extraction. Genotyping laboratories using a low-sensitivity technique should receive paraffin-embedded material containing more than 30% of neoplastic cells.50 ,51 It is noteworthy that the determination of the percentage of tumour cells is very much ‘observer-dependent’. In an external quality assessment review, the same specimen was deemed to contain 10%–20% of tumour cells by one laboratory and 90%–100% by others; hence, the observer variability on a single case can be as high as 80%.52 Depending on the complexity of histology and on the density of the tumour, the DNA extracted from four (resection specimens) or five (biopsy specimens) 3 μm thick serial sections is usually sufficient. The fifth section serves to confirm that sections 2–4 contain tumour tissue. Neutral-buffered formalin is the preferred fixative, although this is not strictly necessary, as shown in countries in which processing of colectomy specimens involves fixation with unbuffered formalin.53 In the absence of a paraffin tissue block, DNA extracted from H&E-stained tissue sections can be used as a starting material.54 The ESP and CAP do not recommend any one single method. In fact, each institution decides whether to validate a laboratory-based assay or to adopt commercial kits.55 The decision is usually based on the equipment, experience and personnel available.52 In figure 1, results from different methods of KRAS mutation detection are reported.

Different methods of KRAS mutation detection, including direct sequencing (A and B), TheraScreen (C and D), pyrosequencing (E and F) and high-resolution melting analysis (G and H). For each method, the top panel (A, C, E and G) shows a wild-type result, while the bottom panel (B, D, F and H) shows (arrows) a mutant (G13D) result.
Many molecular methods are available; all include an initial PCR amplification of the KRAS target sequences. For assays that are not approved for in vitro diagnostic (IVD) use by the European Community or by the US Food and Drug Administration (FDA), the performance characteristics of the assay must be determined and validated by the clinical laboratory before implementation. Kamel-Reid et al56 illustrated how to validate the performance characteristics of KRAS mutation assays by assessing accuracy, precision, analytical sensitivity and specificity, reportable range and reference range. As a general rule, a mutation frequency of 40% and a cluster of three mutation types (p.G12D, p.G12V and p.G13D) in primary tumours and metastases can be considered benchmarks for routine KRAS analyses.57
KRAS mutational status by direct sequencing
Direct sequencing of PCR products is considered a reliable and low-cost standard method for KRAS mutation detection.58 As a general rule, samples featuring tissue areas with more than 30% of neoplastic cells, possibly selected by manual macrodissection,59 ,60 can be reliably tested by direct sequencing. These represent the vast majority of specimens analysed in the routine setting. In fact, in a recent review of 578 cases referred to our laboratory, we found that 528 (91.3%) specimens contained more than 30% of neoplastic cells.61 Besides the percentage of neoplastic cells, the limit of detection (LOD) of this technique partly depends on the specific mutation and on the experience of the person interpreting the data.62 When a KRAS mutation is identified by direct sequencing, both mutant and wild-type alleles are seen on the sequencing electropherograms. In a minority of cases, the electropherogram shows low-intensity peaks that are suggestive of KRAS mutations, but mutations must be verified with a more sensitive technique.63 ,64
In some instances, even if the tumour has not been microdissected, the mutant allele may appear to be in great excess of the wild-type allele.65 The mutant allele may become dominant when deletion of the wild-type allele and/or chromosome 12 hyperploidy or KRAS amplification leads to mutant allele-specific imbalance (MASI).65 ,66 Tumours harbouring extra copies of mutant KRAS alleles can also be identified by pyrosequencing.64 KRAS MASI correlates with a worse overall survival (OS), especially among patients with KRAS codon 13 mutations.66
Identification of KRAS mutational status by high-resolution melting analysis
High-resolution melting analysis (HRMA) is a rapid, highly sensitive and cost-effective, in-tube screening tool. It does not identify the specific mutation present but detects DNA sequence variations based on specific sequence-related melting profiles of PCR products.67–71 Positive results need confirmation,70 which is usually obtained by direct sequencing.68–73 However, in cases of a low concentration of the mutant allele, the results of direct sequencing and the more sensitive HRMA can be discordant.68 ,69 ,72–74 We recently reported that HRMA identified mutations in 4/50 patients that had been missed by direct sequencing.75 Thus, tools more sensitive than direct sequencing are required to confirm positive HRMA samples, and, in routine diagnostics, positive HRMA results may be verified with commercial kits such as the TheraScreen KRAS Mutation Detection Kit (DxS-Qiagen). Ultra-deep pyrosequencing of KRAS amplicons with the 454 GS Junior system was recently found to be cost-effective in confirming HRMA KRAS genotyping.67
Identification of KRAS mutation status with the TheraScreen kit
The TheraScreen KRAS Mutation Detection Kit (DxS-Qiagen), recently approved for IVD by the FDA, detects the seven most frequent somatic mutations in codons 12 and 13. The LOD of mutant alleles is between 1% and 5% depending on DNA quality.53 Each DNA sample is added to eight separate reaction tubes, and Scorpion probes detect fluorescence when the specific primer fully matches the target sequence. The PCR analysis is carried out in less than 2 h, and the presence of a KRAS mutation is scored by threshold cycle cut-off values provided by the kit manufacturer.
This technology has a high analytical efficiency.59 ,76–78 Tol et al60 obtained concordant results with sequencing and the TheraScreen assay in 486/510 (95.3%) samples. The few discrepancies observed reflected the higher sensitivity of TheraScreen in samples with a tumour cell percentage below 30.60 Similarly, in the cohort of 213 patients tested by Dono et al,79 the sequencing and TheraScreen methods were highly concordant (97.6%). Carotenuto et al,63 Pinto et al80 and Franklin et al59 confirmed that the TheraScreen kit is more sensitive than direct sequencing. However, in the series reported by Tol et al,60 4 out of 510 samples (1%) were false negative with the TheraScreen assay because the sequence alterations were not covered by the kit.
Several studies showed that KRAS mutation status assessed by TheraScreen has clinical significance. Indeed, this technology was used in several retrospective analyses that showed the efficacy of anti-EGFR therapy in relation to KRAS status.2 ,6 ,7 ,81 More recently, tumour samples from 394 of the 572 patients enrolled in the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG) CO.17 phase 3 trial were retrospectively tested by the TheraScreen assay, and the data relative to the OS of wild-type patients supported the clinical utility of the kit.61 ,82 ,83
KRAS mutational status by cobas
The cobas KRAS Mutation Test (Roche Molecular Systems) is a robust, accurate TaqMelt real-time PCR test. Fifty nanograms of DNA extracted from a single paraffin section are required.84 The test detects 19 mutations in codons 12, 13 and 61 with an LOD of 5%.85 The accuracy of the test is similar to that of massively parallel pyrosequencing.84 The final result is ‘mutated in codons 12 and 13’ without indication of the exact mutation. However, codon 12 and 13 mutations may differ in terms of their clinical impact. Indeed, evidence derived retrospectively in a small cohort (n=32) of chemotherapy-refractory mCRC patients suggests that patients with tumours harbouring G13D mutations (the third most frequent KRAS mutation in CRC)86 may benefit from anti-EGFR antibody therapy.87 Prescreening with cobas associated with additional TaqMan mutation characterisation is an easy and reliable approach for routine diagnostic purposes.88
KRAS StripAssay
User-friendly test strips can promote the widespread implementation of KRAS testing. The KRAS StripAssay combines mutant-enriched PCR based on peptide nucleic acid clamping and reverse hybridisation of amplification products to nitrocellulose test strips that contain a parallel array of oligonucleotide probes targeting 10 frequent mutations in codons 12 and 13 of the KRAS gene.89 ,90 This assay is more sensitive (analytical sensitivity 1%) than direct sequencing91 and is relatively fast (<6 h excluding DNA isolation). The StripAssay is a practical alternative to direct sequencing when only a few tumour cells are available. However, it is recommended to confirm StripAssay-positive samples using a new StripAssay or another assay that has a similar analytical sensitivity.92
KRAS mutational status by pyrosequencing
Pyrosequencing requires only10 ng of DNA and involves light emission at each position after the incorporation of a nucleotide into the synthesised DNA complementary to the region of interest, which is usually less than 50 nucleotides.93 Various studies have consistently shown that pyrosequencing has an LOD of 5% for mutant alleles.64 ,94 ,95 Both in-house developed assays 95–98 and CE-IVD-marked PyroMark kit (Qiagen)77 have been extensively used to detect KRAS mutations in codons 12, 13 and 61. Pyrosequencing can require confirmatory testing in rare instances, that is, when suboptimal DNA results in a low signal strength.77
Analysis of KRAS mutational status by next-generation sequencing
Thanks to next-generation sequencing (NGS) technologies, it is now possible to screen simultaneously multiple mutations in multiple genes in a single test run. Detection of targeted oncogene mutations, including KRAS mutations, in CRC formalin-fixed, paraffin-embedded specimens by NGS has an accuracy of 96.1% (compared with Sanger sequencing) and 99.6% (compared with real-time PCR methods).99 Accurate quantitative results in mutant allelic frequency can be achieved at a higher throughput scale with KRAS amplicons that are represented in 9 × 103 to 12×103 reads per sample.67 The technical obstacles to the use of NGS in clinical practice are currently being addressed.100 ,101
Practice
KRAS testing is increasingly being used to guide treatment selection for patients with mCRC. In a survey of 14 countries of Europe, Latin America and Asia, the frequency of KRAS testing of mCRC increased from 3% in 2008 to 69% in 2010.8 Knowledge of the patient's KRAS mutation status seems also to influence the choice of the targeted agent. In fact, patients with wild-type KRAS were more frequently treated with cetuximab, while patients with a KRAS mutant tumour or a KRAS unknown mutant tumour were more often treated with bevacizumab.8 Similar data were obtained in a US Community Setting.102 In a more recent observational French study, 433/583 (81.1%) mCRC patients underwent KRAS testing.103 It was mainly requested by oncologists (n=195; 45.5%) and gastroenterologists (n=133; 31.0%).103
The KRAS testing turnaround time is influenced by the sequential involvement of several different health professional rather than by the type of molecular procedure.104 A rational workflow must be established at each site to reduce the time spent reviewing the sample from the pathology archive, reviewing the slides for tumour cells and extracting DNA.64 High-volume testing laboratories have a turnaround time of 10–14 working days.105 Considering the importance of KRAS mutation status for treatment decision making in patients with mCRC, the test result should be available in about seven working days.104 In 2010, KRAS test results were obtained within 15 days in 82%, 51% and 98% of laboratories in Europe, Latin America and Asia, respectively.8 In France, where KRAS testing is routinely performed in a few centralised laboratories, KRAS status becomes available within 23.6±28.2 days for 87% of patients.103
KRAS external quality assessment programmes
To ensure the reliability of KRAS testing, external quality assessment programmes that mirror the daily diagnostic situation have been conducted in Germany,106 Italy,50 the UK,107 North America105 ,108 and in a European programme setup by the ESP.52 The results of the first round of the ESP assessment, conducted in 59 laboratories of 8 European countries, revealed that only 70% of laboratories correctly genotyped all samples.52 Three aspects of molecular testing were assessed: the percentage of neoplastic cells in the specimen, the molecular test itself and reporting, as recommended by van Krieken et al.52 ,109 Because the correct mutation call rate decreases in proportion to the decreasing percentage of tumour cells in a specimen, quality assurance programmes should include samples with a low tumour content.110 To this aim, artificial paraffin blocks consisting of mutation-positive colorectal cancer cells diluted in a background of mutation phase are useful.111 However, this approach does not take account of test variables linked to the preanalytical phase, that is, specimen fixation, dehydration, clearing and embedding.112 Fragmentation of DNA during tissue processing may lead to artifactual KRAS mutations, whose frequency is not negligible (4.7%).21 DNA treatment with Escherichia coli uracil N-glycosylase before amplification and genotyping on shorter amplicons may be a way to avoid artifactual mutations.21
Genetic intratumoural heterogeneity and treatment outcome
The tissue distribution of KRAS mutant cells is homogeneous in most colorectal carcinomas.113 Goranova et al114 analysed multiple tumour areas by laser capture microdissection and found that a single dominant clone occupied approximately 80%–90% of the tumour volume. However, heterogeneity is not negligible.115 Indeed, Richman et al115 described a 7% discordance among tumour blocks, whereas Baldus et al reported a 8% discordance between the tumour centre and the invasion fronts.113 Heterogeneous KRAS status has been reported in 11.6% of primary tumours.116
The threshold level of KRAS-mutated cells within a tumour mass that is resistant to cetuximab treatment is uncertain. Retrospective analyses have been carried out to determine whether cetuximab treatment is effective in tumours harbouring a small number of mutated cells.75 ,79 ,82 ,117 ,118 We used HRMA to look for KRAS mutations in 50 mCRC patients previously found to be KRAS wild type by direct sequencing and treated in a second-line or third-line setting with cetuximab-based therapy.113 ,114 HRMA identified mutations in 4/50 patients that had been missed by direct sequencing. None of these four patients responded to cetuximab treatment, and their progression-free survival (PFS) and OS were very short. Thus, if patient management had been based on HRMA results, a significant percentage (8%) of patients would have been spared useless treatment.75 In the study by Bando et al, the TheraScreen method revealed 9% more KRAS mutations than did direct sequencing. Among the 47 patients with complete clinical information who were wild type by direct sequencing and had been treated with cetuximab alone or combined with irinotecan, the 9 patients found mutated by TheraScreen failed to respond and had a significantly shorter PFS and OS than TheraScreen wild-type patients.82 Molinari et al identified mutations using the highly sensitive mutant-enriched PCR (eME-PCR) method in 55/111 patients (49.5%), whereas the mutation rate in exon 2 by direct sequencing was 43/111 (38.7%).117 None of the 12 patients with a KRAS mutant at eME-PCR responded to anti-EGFR monoclonal antibody-containing therapy.117 Similarly, in the study by Dono et al,79 26/32 (82.2%) patients initially considered KRAS wild type and reclassified as KRAS mutated with locked nucleic acid PCR failed to respond to anti-EGFR therapies. Kimura et al119 obtained similar results with a high-sensitivity two-step PCR restriction fragmentation length polymorphism method. Differently, using pyrosequencing, Santini et al118 detected KRAS mutations in 3/29 patients (10.3%) previously identified as KRAS wild type by real-time PCR using allele-specific oligonucleotide primers. However, these three patients responded to treatment with cetuximab combined with irinotecan.118
The above contrasting results may be due to the limited number of cases analysed, the different populations of patients (one, two or more previous lines of treatment for metastatic disease), different treatment regimens (anti-EGFR monoclonal antibody alone or in combination with chemotherapy) and different mutation detection panels.
The future
Only a subset of mCRC patients selected by KRAS testing benefit from EGFR-targeted therapy. Thus, there is a need for studies aimed at identifying additional genetic determinants of primary resistance. Thus far, only negative predictors have been investigated, mostly in retrospective analyses. An increase in KRAS gene copy number (GCN) has been associated with a more active ‘mutation-like’ phenotype.120 Smith et al showed, by Taqman-based and fluorescence in situ hybridisation (FISH) analyses, that KRAS GCN is increased in a small subset (2%) of wild-type tumours. Valtorta et al121 confirmed that this event is rare (0.67%) and mutually exclusive with KRAS mutations. A study based on KRAS CGN and microRNAs suggested that patients carrying a high CGN of wild-type KRAS may not respond to cetuximab administration.122 Interestingly, a high CGN of wild-type KRAS may also be acquired during treatment with EGFR inhibitors.123
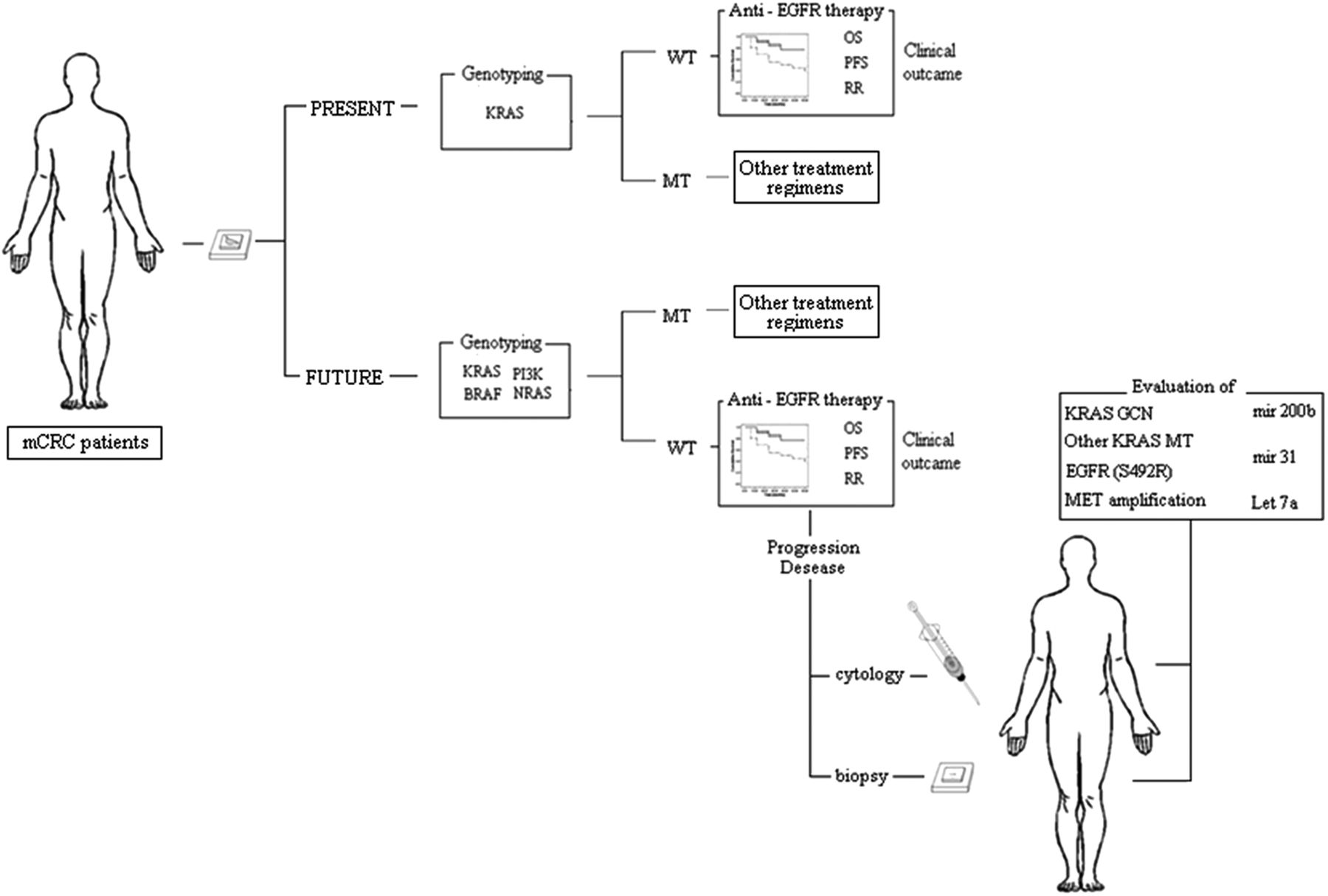
To date, only KRAS testing has been implemented in clinical practice. The tumours of patients with mCRC are only profiled for seven KRAS mutations before receiving cetuximab or panitumumab.51 However, it is conceivable that, in the not too distant future, the comprehensive integrated analysis of the KRAS–RAF–MAPK and the PI3K–PTEN–AKT signalling pathways will enable us to identify most of the mCRC patients who are unlikely to respond to anti-EGFR therapies (figure 2). BRAF is the principal effector of KRAS. Although the presence of BRAF in its wild-type form is required for response to treatment,124 BRAF mutations define a genetically distinct subset of CRCs characterised by an extremely poor prognosis.125 Sartore-Bianchi et al126 proposed that CRCs lacking alterations in KRAS, BRAF, PTEN and PIK3CA be defined as ‘quadruple-negative’. Notably, molecular alterations such as BRAF and PIK3CA (exon 20) mutations can co-occur in a single tumour.126 Other panels include the assessment of Neuroblastoma RAS viral oncogene homolog (NRAS) mutations.125 Approximately 20% of quadruple-negative CRC patients do not respond to anti-EGFR-targeted therapies, suggesting that genotyping-based selection of patients without KRAS, BRAF, NRAS and PIK3CA mutations for treatment with cetuximab is not sufficient and, consequently, that the mechanisms underlying alterations in other key elements of the EGFR-dependent signal cascade need to be unravelled.111
{kind=link}
{kind=link}
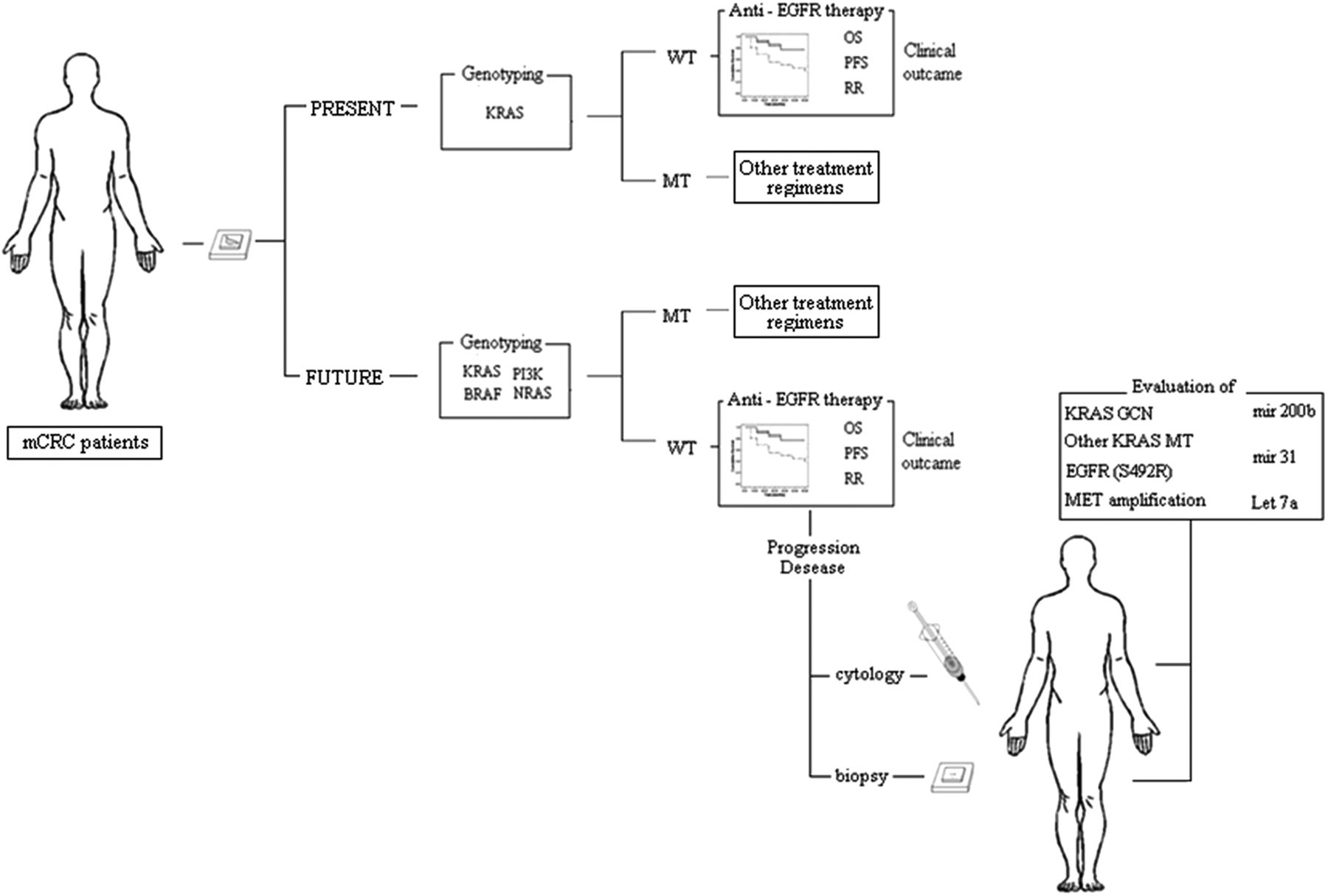
Metastatic colorectal cancer (mCRC) patient triage to anti-epidermal growth factor receptor (EGFR) treatment in clinical practice: present and future prospectives. WT, wild type; MT, mutant; OS, overall survival; PFS, progression-free survival; RR, response rate.
The clinical efficacy of anti-EGFR cancer therapies is limited by the inevitable development of acquired drug resistance. In the last few years, various mechanisms of resistance have been defined and drugs to tackle the resistant cells are being developed.127 Bouchahda et al128 reported the appearance of a KRAS mutation during the course of mCRC in a patient who initially carried wild-type KRAS. In a recent study, preclinical models and patients’ samples were evaluated to determine whether KRAS mutation and/or amplification are clinically relevant mechanisms of acquired cetuximab resistance.123 Recently, deep sequencing showed that 6 post-treatment tumour biopsies from 10 patients with mCRC who had become refractory to anti-EGFR therapy were mutated, including a case mutated outside codons 12 and 13 (Q61H), whereas the matched pretreatment biopsies were negative.123 Prospective clinical trials with serial assessments of KRAS status during anti-EGFR treatment are required to define the frequency of KRAS mutations as a mechanism of acquired resistance to anti-EGFR therapies.127 Resistance can also result from mutations in the EGFR, which is the drug target itself. Montagut et al129 identified an acquired EGFR ectodomain mutation (S492R) that prevents cetuximab binding and activity. However, tumours carrying the EGFR S492R mutation may still be effectively treated with panitumumab (figure 2). The development of prospective clinical trials for cetuximab-resistant individuals harbouring this mutation will shed light on the response rates to panitumumab administration in this setting. Very recent evidence suggests that the amplification of the mesenchymal-epithelial transition factor (MET) proto-oncogene is associated with acquired resistance in KRAS wild-type tumours.130 Prospective clinical trials designed to assess the activity of MET inhibitors in patients displaying resistance as a result of METamplification are required.
Several microRNAs implicated in KRAS regulation may have predictive value.131 Elevated expression of miR-200b 122 or of Let-7a132 in the presence of KRAS mutation reduces KRAS protein levels and improves clinical outcome in patients treated with cetuximab. In the absence of KRAS and BRAF mutations, increased miR-31 and decreased miR-592 expression were associated with poor response to treatment.133 In CRC KRAS wild-type patients, low miRNA-143 expression in tumour tissue is an independent negative prognostic factor, but it is not predictive of the response to EGFR-targeted agents.134
Conclusions
Testing tumour tissue for predictive gene mutations to guide personalised therapy is a rapidly emerging field in pathology. Standardising molecular testing and harmonising molecular pathology with traditional histopathology are challenging. The implementation of KRAS mutation testing reinforces the key function played by the surgical pathologist in the multidisciplinary management of CRC.
Such local issues as equipment, expertise and personnel available have led to different approaches to KRAS mutation testing. In fact, the test is still poorly standardised. Besides the variables linked to preanalytical tissue processing, molecular testing can be carried out in-house or in centralised laboratories on different types of samples (resection, biopsy and cytological slides) from different sources (primary and/or metastatic) with laboratory-based assays or with commercial kits using a wide range of techniques, each of which differ in performance. However, there are far more crucial issues involved in KRAS testing: the DNA sample should be representative of a sizeable number of CRC cells; the percentage of mutated alleles should be within the analytical sensitivity range of the method used; the laboratory should validate the performance characteristics of the KRAS mutation analysis used; and last but not least, the laboratory should undergo internal and external quality assessments.
Investigations are required to clarify the relationship between KRAS genetic intratumoural heterogeneity and clinical sensitivity of mutational detection tools in relation to anti-EGFR treatment outcome. The comprehensive integrated analysis of multiple biomarkers and the serial assessments of KRAS status during anti-EGFR treatment will help to select more accurately patients with primary or acquired resistance to anti-EGFR therapy.
Key messages
-
The DNA sample should be representative of a sizeable number of CRC cells.
-
The percentage of mutated alleles should be within the analytical sensitivity range of the method used.
-
The laboratory should validate the performance characteristics of the KRAS mutation analysis used.
-
The laboratory should undergo internal and external quality assessments.
Acknowledgments
We are grateful to Jean Ann Gilder (Scientific Communication srl) for text editing.
References
Footnotes
UM and CC contributed equally.
-
Contributors UM, CC, CdL, CB and GT reviewed literature data. UM evaluated the technical issues. CC evaluated the clinical issues. GT wrote the manuscript and is the guarantor of the study.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.