Article Text

Abstract
Aims Adrenocortical carcinoma (ACC) carries a poor prognosis and current systemic cytotoxic therapies result in only modest improvement in overall survival. In this retrospective study, we performed a comprehensive genomic profiling of 29 consecutive ACC samples to identify potential targets of therapy not currently searched for in routine clinical practice.
Methods DNA from 29 ACC was sequenced to high, uniform coverage (Illumina HiSeq) and analysed for genomic alterations (GAs).
Results At least one GA was found in 22 (76%) ACC (mean 2.6 alterations per ACC). The most frequent GAs were in TP53 (34%), NF1 (14%), CDKN2A (14%), MEN1 (14%), CTNNB1 (10%) and ATM (10%). APC, CCND2, CDK4, DAXX, DNMT3A, KDM5C, LRP1B, MSH2 and RB1 were each altered in two cases (7%) and EGFR, ERBB4, KRAS, MDM2, NRAS, PDGFRB, PIK3CA, PTEN and PTCH1 were each altered in a single case (3%). In 17 (59%) of ACC, at least one GA was associated with an available therapeutic or a mechanism-based clinical trial.
Conclusions Next-generation sequencing can discover targets of therapy for relapsed and metastatic ACC and shows promise to improve outcomes for this aggressive form of cancer.
- Cancer Genetics
- Endocrine Pathology
- Molecular Pathology
- Oncology
- Gene Amplification
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Adrenocortical carcinoma (ACC) is a primary malignant neoplasm of the adrenal cortex that can vary widely in histologic appearance.1–3 ACC can occur at any age and has an annual incidence of 0.7–2.0 cases per million people with a peak incidence between 40 and 50 years (1–3). The disease occurs in women more often than men by a ratio of up to 1.5 to 1.1–3 ACC occurs both as an inherited form of cancer which is particularly prominent in some populations and also as a sporadic tumour.1–3 The most common inherited predisposition is associated with the Li-Fraumeni Syndrome (LFS) and germline TP53 mutations, but the disease has also been consistently linked to the Lynch Syndrome and germline alterations in DNA repair genes.4–6 In children, TP53 germline mutations may be present in 50–80% of ACC cases, whereas in adults, at least 95% of the tumours arise in the absence of germline TP53 alterations.4 ,5 ACC is an aggressive form of cancer and tumours that cannot be completely resected have a particularly poor prognosis.7 Increased immunostaining (IHC) for the cell cycle protein Ki-67 has generally been accepted as the most reliable slide-based biomarker of ACC prognosis.2 ,8 Successful surgical resection for early stage disease is the only known curative procedure for ACC with a 5-year disease-free survival for a complete resection of a Stage I–III tumour of 30%.1–3 For patients with recurrent and metastatic disease, the 5-year relative survival is poor at only 7%.1–3 The selection of medical treatment of ACC has been based on non-randomised trials or retrospective analyses.1–3 The adrenotoxic drug mitotane has been the cornerstone drug for ACC, both in adjuvant and metastatic disease settings with a recent emergence of platin-based regimens.9 ,10 Recently, the addition of three cytotoxic drugs, etopiside, doxorubicin and cisplatin to mitotane showed improvement in response rates and disease-free progression for metastatic ACC.11 Thus, given the poor prognosis of recurrent and metastatic ACC patients treated by chemotherapy, there has been emerging interest in studying whether comprehensive DNA sequencing of ACC tumours using next-generation sequencing (NGS)-based genomic profiling could detect genomic alterations (GAs) that could be used to guide targeted therapies for the personalised treatment of this challenging disease.
Methods
Genomic profiling was performed in a CLIA-certified, CAP-accredited reference laboratory (Foundation Medicine). DNA extracted from clinical formalin-fixed paraffin-embedded tumour samples of 29 consecutively submitted ACC samples was analysed by hybridisation capture of 3320 exons from 236 cancer-related genes and 47 introns of 19 genes commonly rearranged in cancer. At least 50 ng of DNA per specimen was isolated and sequenced to high, uniform coverage (mean 734X) on the Illumina HiSeq2500 instrument, as previously described.12 GAs (base substitutions, short insertions and deletions, focal gene amplifications, homozygous deletions and select rearrangements) were determined and then reported for each patient sample. To maximise mutation detection sensitivity in heterogeneous ACC biopsies and resections, the test was validated to detect base substitutions at ≥10% mutant allele frequency with ≥99% sensitivity and indels at ≥20% mutant allele frequency with ≥95% sensitivity, with a false discovery rate of <1%.12 Actionable alterations are defined as those whose effect is targetable using anticancer drugs currently on the market or in registered clinical trials. Local site permissions to use clinical samples were obtained for this study.
Patient characteristics are shown in table 1. All 29 (100%) patients had developed locally advanced and/or metastatic ACC refractory to their last line of cytotoxic chemotherapy. The median age of the ACC patients at the time of sequencing was 50 years (range 21–74 years). There were 12 (41%) ACC with predominantly oncocytic tumour cell cytoplasm, 12 (41%) ACC with markedly pleomorphic nuclei and 5 (17%) ACC with predominantly clear cell cytoplasm. There were 11 (40%) Grade 3 and 18 (60%) Grade 4 tumours using the Fuhrman grading system (table 1). Two ACC were Stage II, 4 were Stage III and 23 were Stage IV at the time of profiling. Sequencing was performed on the original primary ACC in 11(38%) and on a metastasis or recurrence biopsy or resection in 18 (62%) ACC (table 1).
Clinicopathologic features of the 29 cases of adrenocortical carcinoma
Results
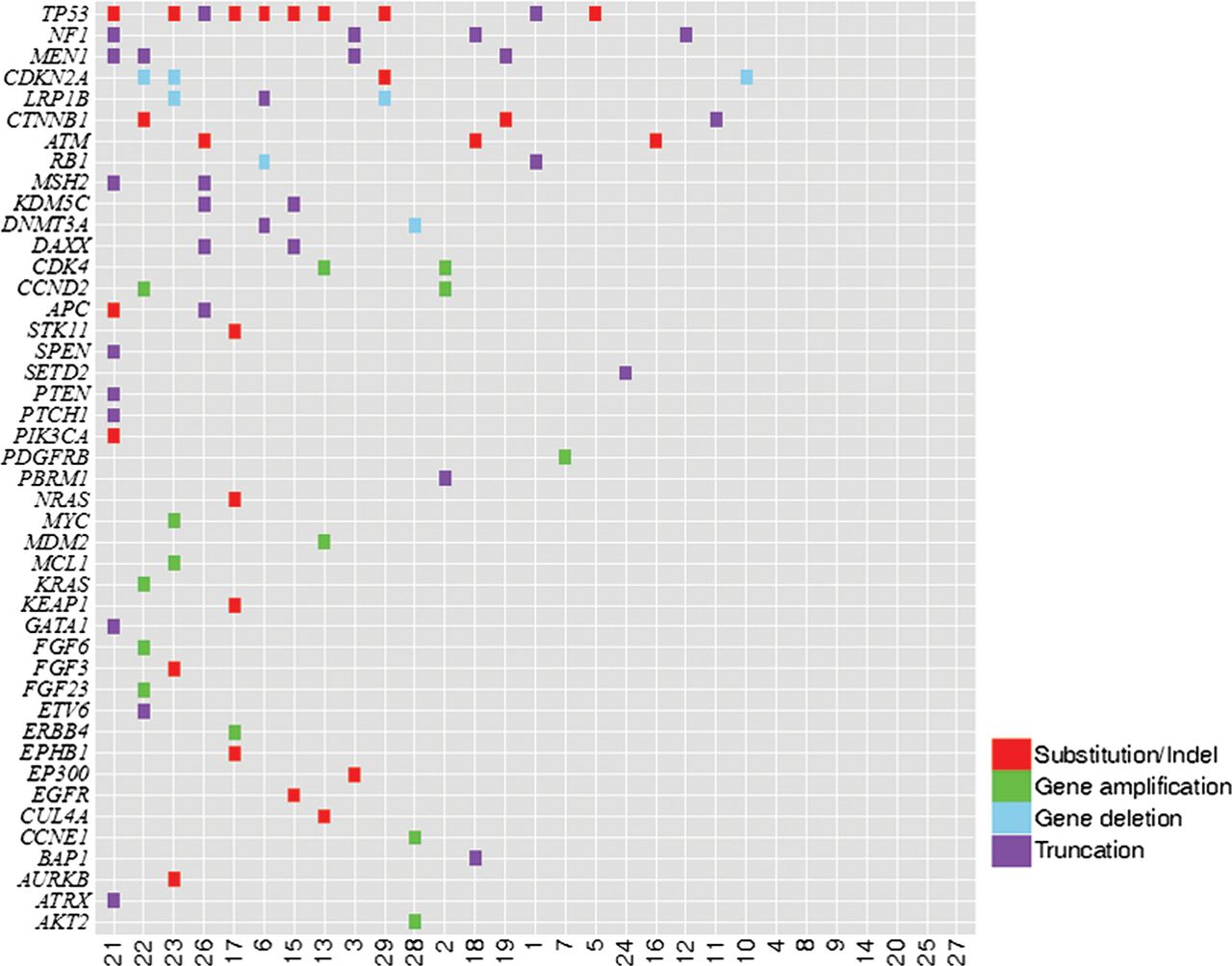
A total of 76 alterations were identified (25 base substitutions and short indels, 14 gene amplifications, 7 gene homozygous deletions and 30 gene truncations) in 43 genes, with 22 cases (76%) harbouring at least one alteration, for a mean of 2.6 alterations per tumour (table 2, figure 1). No gene fusions were identified. The most common biologically relevant alterations that cannot currently be linked to a targeted treatment option were found in TP53 (34%), MEN1 (14%) CTNNB1 (10%), APC (7%), DAXX (7%), KDM5C (7%), LRP1B (7%), MSH2 (7%) and RB1 (7%). At least one clinically meaningful alteration that could potentially guide decisions for targeted treatment was found in 59% (17/29) of the ACC cases. The most common potentially actionable alterations involved NF1 (14%), CDKN2A (14%), ATM (10%), CCND2 (7%), CDK4 (7%), DNMT3A (7%) with EGFR, ERBB4, KRAS, MDM2, NRAS, PDGFRB, PIK3CA, PTEN, PTCH1 and STK11, each altered in a single case. There were no observable differences in the pattern of GAs of the ACC where the primary tumour was sequenced compared with ACC where a metastasis sample was used.
Genomic alterations (GAs) identified in 29 cases of adrenocortical carcinoma (ACC)

Tile plot of genomic alterations identified in 29 adrenocortical carcinoma cases.
Discussion
The genetic background and pathogenesis of ACC have been widely studied using a variety of methodologies.13–16 Recent molecular analysis of ACC has predominantly studied DNA copy number alterations by comparative genomic hybridisation, mRNA levels by gene expression profiling and epigenetic alterations by PCR-based methods.13–22 TP53 mutations have been reported in ACC at frequencies ranging from 10% to 70% and have been associated with decreased disease-free survival and poor outcomes.23–26 Germline TP53 mutations have also been linked with the development of ACC, particularly in paediatric patients with a family history of LFS and Li-Fraumeni-like Syndrome.26 In this study, the mean age of the patients with ACC with and without TP53 mutation was 59.4 years versus 50.2 years, respectively, and no TP53-mutated ACC was identified in a patient younger than 46 years. Patient-matched normal specimens required to definitively determine the germline status of TP53 mutation were not available for this study.
Given the limited success of systemic cytotoxic chemotherapy in the treatment of relapsed and metastatic ACC, investigators have queried whether genomic profiling could uncover potential targets of therapy not routinely searched for in the management of this disease.27 ,28 In one previous NGS-based study of ACC interrogating fewer genes and using a method that does not detect all classes of GAs, a few obvious genomic-derived therapeutic targets emerged.29 In the current study, interrogating an expanded series of cancer-related genes with an assay capable of detecting all classes of GAs, half of the ACC harboured potentially clinically meaningful GAs.12
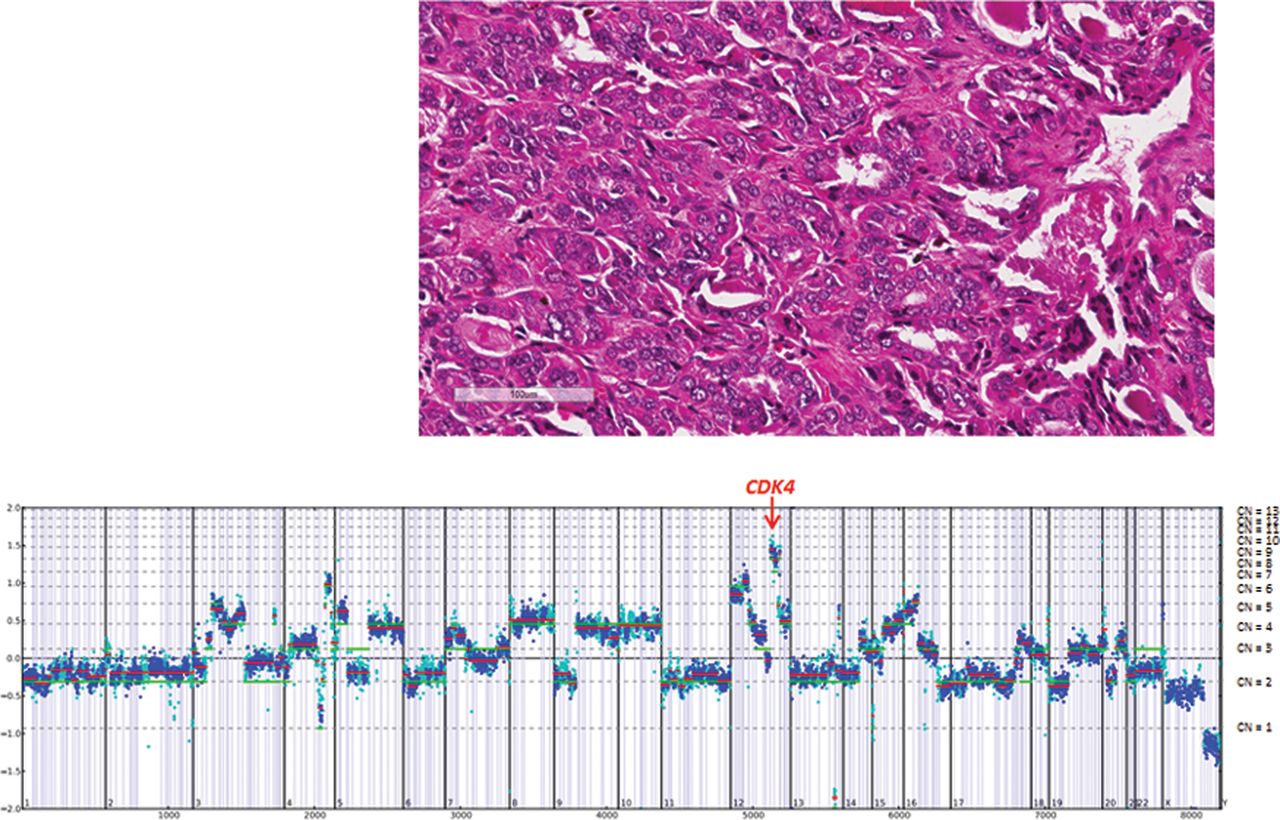
For example in Case 13, a pleomorphic ACC derived from a 48-year-old man that had metastasized to the liver, amplifications of CDK4 and MDM2 were identified (figure 2). CDK4 encodes cyclin-dependent kinase 4, which, along with functional homologue CDK6 and family member CDK2, regulates cell cycle G1 phase progression and the G1/S transition.30 Amplification of CDK4 has been identified in multiple cancer types and in a small number of adrenal carcinomas.31 A number of drugs that target CDK4 are under investigation in phase I clinical trials.32 Similarly, therapies targeting MDM2 are under study in clinical trials.33 In another case (Case 7), a locally advanced oncocytic ACC derived from a 60-year-old man, a single alteration was identified, amplification of PDGFRB (figure 3). GAs, including amplification of PDGFRB, have not been reported in ACC in the literature until now. PDGFRB amplification has been associated with PDGFRB protein overexpression and increased kinase activity in a variety of other tumours.34 Although there are no PDGFRB inhibitors currently approved for use in ACC, several drugs that inhibit PDGFRB, including dasatinib, imatinib, sorafenib and sunitinib, have been food and drug administration (FDA) approved for use in other tumour types. Although an initial study indicated significant efficacy of sorafenib in the treatment of metastatic ACC, more recent phase II trial was unable to duplicate that result.35 ,36 The vascular endothelial growth factor (VEGF) inhibitor axitinib has also shown limited impact on the outcome of this disease.37
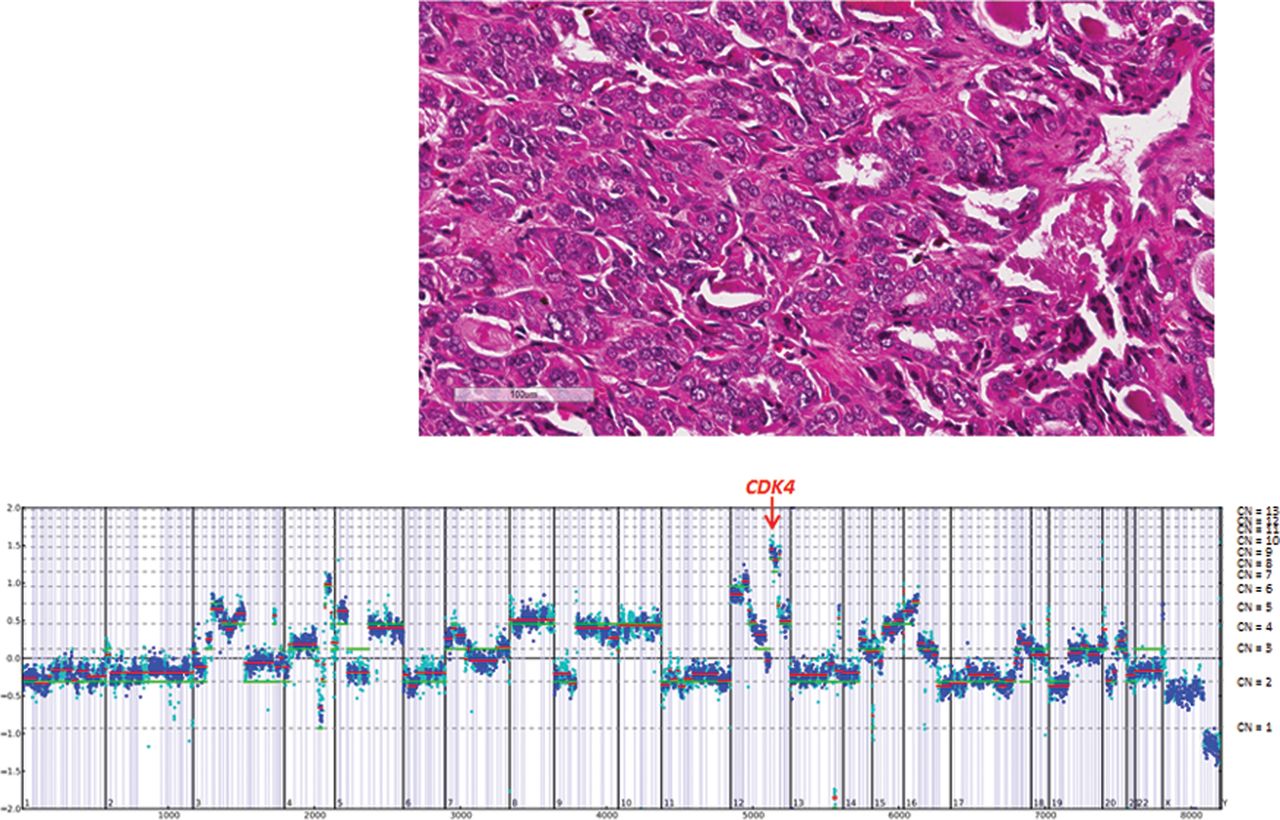
Case 13. A pleomorphic adrenocortical carcinoma liver metastasis derived from a 48-year-old man harboured CDK4 and MDM2 amplifications, CUL4A (V275M) and TP53 (S241Y) mutations.

{kind=link}
{kind=link}
{kind=link}
Case 7. A Stage III Fuhrman Grade 3 adrenocortical carcinoma with extensive tumour necrosis derived from a 60-year-old man harboured PDGFRB amplification.
The poor prognosis of patients with locally advanced and metastatic ACC has increased interest in identifying targeted therapies for patients with ACC.38–40 The GAs identified linked to targeted therapies in this study involved multiple genes and pathways, including the receptor tyrosine kinase, retrovirus associated sarcoma (RAS) signalling, molecular targets of rapamycin (MTOR) and Hedgehog pathways. These alterations are thus candidates for referral of patients to a broad series of mechanism-driven registered clinical trials using targeted therapies such as MTOR inhibitors, epidermal growth factor receptor (EGFR) inhibitors, Hedgehog pathway inhibitors and cyclin dependent kinase (CDK) inhibitors.
A recent study further classified ACC based on genomic signatures into indolent and aggressive subtypes.41 This study reported many, but not all of the alterations found in the current study which was restricted to aggressive ACC which had relapsed locally or spread to regional or distant sites. In this study, clinically meaningful GAs that could potentially guide targeted treatment options were identified in 58% of patients sequenced (table 3). However, the complexity of the alterations identified presents challenges to the NGS platform used to test patients with relapsed ACC resistant to conventional therapies. The use of NGS to discover novel targets of therapy not routinely searched for in the current management of ACC shows promise and warrants further study and the development of new mechanism-driven clinical trials designed to improve outcomes for this aggressive form of cancer.
Significant targetable genomic alterations (GAs) discovered by NGS assessment of 29 cases of adrenocortical carcinoma (ACC)
Take home messages
-
A sensitive/validated next-generation sequencing (NGS) assay can readily be performed on formalin-fixed paraffin-embedded biopsies of patients diagnosed with adrenocortical carcinoma (ACC).
-
In 22 (76%) ACC, at least one genomic alteration (GA) was identified (mean 2.6 alterations per ACC).
-
The most frequent GAs in ACC were in TP53 (34%), NF1 (14%), CDKN2A (14%), MEN1 (14%), CTNNB1 (10%) and ATM (10%). APC, CCND2, CDK4, DAXX, DNMT3A, KDM5C, LRP1B, MSH2 and RB1 were each altered in two cases (7%) and EGFR, ERBB4, KRAS, MDM2, NRAS, PDGFRB, PIK3CA, PTEN and PTCH1 were each altered in a single case (3%).
-
In 17 (59%) ACC, at least one GA was associated with an available therapeutic or a mechanism-based clinical trial.
-
NGS can discover targets of therapy for relapsed and metastatic ACC and shows promise to improve outcomes for this aggressive form of cancer.
References
Footnotes
-
Contributors The following authors contributed to the development and submission of this manuscript. Substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work: JSR, KW, JVR, MJP, CES, SA, JAE, EL, CH, JB, GAO, RY, DL, DM, JC, VAM, PJS. Drafting the work or revising it critically for important intellectual content: JSR, KW, JVR, MJP, CEES, SMA, JAE, ELB, CH, JB, GAO, RY, DL, DM, J, VAM, PJS. Final approval of the version to be published: JSR, KW, JVR, MJP, CES, SMA, JE, ELB, CH, JB, GAO, RY, DL, DM, JC, VAM, PJS. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: JSR, KW , JR, MJP, CES, SMA, JAE, ELB, CH, JB, GAO, RY , DL, DM, JC, VAM, PJS.
-
Competing interests None.
-
Ethics approval Local site permissions were used that included signed authorisations from submitting institutions.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All relevant data from this study are included in the submission. A supplementary file, including genomic variants of undetermined significance not reported in patient records, can be prepared and sent to reviewers if required. This file would then be made available to the readership.