Article Text

Abstract
Analysis of colorectal carcinoma (CRC) tissue for KRAS codon 12 or 13 mutations to guide use of anti-epidermal growth factor receptor (EGFR) therapy is now considered mandatory in the UK. The scope of this practice has been recently extended because of data indicating that NRAS mutations and additional KRAS mutations also predict for poor response to anti-EGFR therapy. The following document provides guidance on RAS (i.e., KRAS and NRAS) testing of CRC tissue in the setting of personalised medicine within the UK and particularly within the NHS. This guidance covers issues related to case selection, preanalytical aspects, analysis and interpretation of such RAS testing.
- COLORECTAL CANCER
- GENETICS
- LABORATORY MANAGEMENT
Statistics from Altmetric.com
Introduction
Colorectal carcinoma (CRC) is the fourth most commonly diagnosed malignancy in the UK with over 40 000 cases diagnosed every year and a 5 year survival rate of 55%.1 In the UK, metastases are demonstrated in 23% of CRC patients with available pretreatment staging data.2 At least 15% of patients who have undergone surgical excision of their tumours will develop recurrent metastatic disease.3 A major step in the treatment of metastatic CRC has been the development of monoclonal antibodies that target and inhibit epidermal growth factor receptor (EGFR). These monoclonal therapies include cetuximab, bevacizumab and panitumumab. There are now consistent data that anti-EGFR therapy is ineffective in CRC with KRAS mutations; much of the earlier data focused on mutations in KRAS codons 12 and 13, which were found to predict for resistance to cetuximab.4 ,5 This finding was embraced by the National Institute for Health and Care Excellence (NICE) in its recommendations on the use of cetuximab for metastatic colorectal carcinoma with liver metastases only (Technology Appraisal 176), that is, analysis of these two KRAS codons should be performed to determine whether a patient is eligible for cetuximab therapy in this specific clinical setting.6 At the time of writing of this document, NICE had not recommended use of EGFR inhibitors for patients with metastatic CRC who have progressed after first-line chemotherapy,7 though funding for such use has been available through the Cancer Drugs Fund (CDF) in England. Therefore, whether use of EGFR inhibitors is being funded by NICE or through the CDF, KRAS genotyping of CRC tissue has become commonly requested within the NHS to help stratify patients for anti-EGFR therapy. Groups outside the UK have already issued guidance or recommendation documents on KRAS testing of CRC.8–10 However, the following document is directed specifically at practice within the UK and especially within the NHS. Further, this guidance is one of the first to incorporate recent data on NRAS testing of CRC in the setting of personalised medicine. The document also reviews some technical and/or investigational aspects that impact directly on RAS testing of CRC. As a document that focuses particularly on practical aspects of such testing, this document is structured to follow the specimen pathway of RAS testing. The main recommendations of this document are summarised in box 1.
Main recommendations for RAS testing of colorectal carcinoma to guide anti-EGFR therapy
-
Network arrangements should be established to ensure rapid and robust tissue pathways from referral centres to testing laboratories.
-
Either primary or metastatic CRC tissue can be used for RAS testing.
-
Either biopsy or resection specimen tissue can be used for RAS testing, though if both are equally available, use of resection tissue is preferable.
-
The minimum neoplastic cell content tested should be at least two times the assay's LOD.
-
RAS analysis should include at least KRAS codons 12, 13, 59, 61, 117 and 146 and NRAS codons 12, 13, 59 and 61.
-
Turnaround time for RAS testing (of the above panel) should be ≤7 working days from receipt of the specimen in the testing laboratory to issuing of the final report, for >90% of specimens.
-
Validation (or verification, where more applicable) of RAS testing assays should be performed and recorded prior to implementation in clinical use.
-
The minimum controls needed for RAS testing should be mutant, wild-type and non-template controls for each region/amplicon analysed.
-
Laboratories should audit their results to ensure that the proportion of mutant cases for each gene and codon are in line with published data. If a significant deviation is seen, the performance of the assay (from specimen reception to reporting of results) should be investigated.
-
Laboratories should make every possible effort to reduce failure rates, including reviewing the quantity and quality of DNA obtained from routine specimens.
-
Laboratories providing RAS testing of CRC should demonstrate successful participation in a relevant EQA scheme, and be appropriately accredited.
Case selection for testing
Reflex or on-demand testing
It remains controversial whether RAS testing of CRC is better practised as a ‘reflex’ or an ‘on-demand’ process. One model of reflex testing requires all surgically excised CRCs to be RAS genotyped and these data integrated into the resection specimen pathology report. These molecular data would therefore be immediately available should the patient be considered for anti-EGFR therapy in the future. This reflex testing would help address the main delay incurred in KRAS testing of CRC in the UK; this delay, in our experience, is not the assay turnaround time once tissue is received by the testing laboratory, but the time required to retrieve an appropriate tissue block and to deliver it to the testing laboratory. Reflex testing may also avoid a potential inability to test a tissue block in the future due to loss of the block, tissue destruction due to suboptimal storage conditions, and/or depletion of the tissue from other uses, for example, research.
A disadvantage of reflex testing is the possibility of unnecessary testing of CRC tissue from patients who never developed metastases. One way to reduce this unnecessary testing is to only perform reflex testing on resected CRCs showing high-risk features for future metastases, for example, those with extramural vascular invasion, nodal metastases and/or a pT4 stage. However, a major disadvantage of reflex testing has been exposed by the emergence of the recent data on RAS mutations beyond KRAS codons 12 and 13 (see below). CRC cases which had already been reflex tested for KRAS codons 12, 13 and perhaps 61, will need to be retested for NRAS mutations and additional KRAS mutations if the patients are now being considered for anti-EGFR therapy. Further, similar scenarios are bound to recur as more genes are found to help predict for resistance to anti-EGFR therapy, and/or as new targeted therapies become available. On-demand testing will most commonly be led by the decision of a relevant multidisciplinary team after consideration of the patient for systemic treatment. At the time of writing of this document, UK laboratories were being reimbursed for RAS testing of only patients with metastatic CRC, which meant that on-demand testing has become the mainstay practice in the UK. To help future-proof this RAS testing service within the UK, resources should be directed to establish robust nationwide systems to improve the tissue pathways for CRC, and to ensure the timely delivery of tissue to testing laboratories for on-demand testing.
Primary or metastatic tissue
Many studies have investigated the concordance of KRAS mutations between primary and metastatic CRC tissues.11 A recent meta-analysis indicates such concordance to be very high: overall rate of 94.1% (95% CIs: 88.3%–95.0%).11 However, there are some data suggesting that there is anatomical variation in such concordance, with lung and nodal metastases showing less genetic concordance with their primary tumours.11 ,12 For example, the above meta-analysis documented an overall concordance rate between nodal metastases and primary CRC to be 81.3% (95% CIs: 69.6%–97.4%).11 Given the absence of absolute genetic concordance between primary tumour and metastasis, if metastatic CRC tissue is available and this tissue can be delivered to the testing laboratory as quickly as primary tumour tissue, testing of the metastatic tissue is preferred. If metastatic tissue is not readily available, the patient's primary tumour tissue should be tested as there is as yet insufficient evidence to warrant biopsy of a metastatic deposit of CRC specifically for RAS genotyping.
Specimen type
A whole range of histopathological or cytological specimens can be used for RAS testing.13 This includes, from our own experience, use of archival H&E or immunostained sections of CRC.
Only a proportion of a tumour is used for testing. In some specimens, such as endoscopic biopsies or core biopsies, this limitation is imposed by the sampling technique. In the case of resection specimens, there are frequently multiple tumour blocks available, but usually one representative block is chosen for testing. Limited sampling, therefore, raises some issues for consideration.
Biopsy containing adenoma only
The first issue involves a patient with clear-cut clinical and radiological evidence of CRC but whose endoscopic tumour biopsies show only adenoma (and no carcinoma), and represent the only tissue available for RAS testing. This is a controversial issue and subsequently is addressed by the authors of this document in different ways. Some feel that the adenoma tissue should not be tested, and repeat biopsy of either the primary tumour or a metastasis should be requested. Other authors feel that such adenoma tissue should be tested and reported if a mutation is identified, but considered to be ‘inadequate’ if no mutation is demonstrated and a repeat biopsy requested. This second approach assumes that because RAS mutations generally occur early on in the adenoma-carcinoma pathway and are accepted to be key driver mutations,14 a RAS mutation demonstrated in an adenoma is likely to be harboured by the CRC arising from the adenoma. However, RAS mutation may also potentially occur as a late event in a CRC,15 and it is therefore possible that an adenocarcinoma may be mutant for RAS while its precursor adenoma is RAS wild-type. A third approach favoured by other authors is similar to the second but only tests adenoma tissue if it shows high-grade dysplasia, this being a more advanced stage in the adenoma-carcinoma sequence.15
Intratumour mutation heterogeneity
Another issue arising from RAS testing of endoscopic biopsies also relates to which of multiple available resected CRC blocks should be tested. This issue focuses on ‘intratumour mutation heterogeneity’, that is, the presence of different RAS genotypes among clones of the same CRC. Endoscopic biopsies represent a limited and superficial sampling of a CRC, and it is possible that mutant clones will not sampled especially if they lie within the deeper parts of the carcinoma. Similarly with resection specimens, a mutant clone may potentially be present in a block which has not been chosen for testing. Based on the most recent KRAS and NRAS data (see below), there is growing consensus that the presence of any one RAS mutation is sufficient to predict for resistance to anti-EGFR therapy. Therefore, mutation heterogeneity with coexistence of more than one RAS mutant in the same CRC is unlikely to have clinical significance. Of potentially more clinical relevance is a combination of some CRC clones being RAS wild-type, and others harbouring a RAS mutation, particularly when the mutant clones represent a minority. This latter scenario is hereafter referred to as ‘low-level mutation’ and, as a reason for a small proportion of RAS mutant allele in a CRC DNA extract, needs to be distinguished from dilution of mutant allele by non-neoplastic DNA in a block with a low neoplastic cell content (see below).
There are some data showing variation of KRAS genotype within a CRC.16 ,17 However, only a minority of CRCs show a mixture of wild-type and KRAS exon 2 or 3 mutant clones (e.g., 13 of 42 CRCs in one study18), and when present, the extent of this genotypic variation is only minor: a KRAS mutant clone occupied >80% of the tumour area for 10 of the 13 abovementioned CRCs.18 Further, a recent study used high-resolution melting curve (HRM) analysis to screen for KRAS mutations in 30 cases of paired endoscopic biopsies and subsequently resected CRCs, and found complete concordance of KRAS genotype (codons 12, 13, 61 and 146) within each pair of specimens.15
Several groups have retrospectively analysed CRCs using more sensitive assays to see whether apparently wild-type tumours harboured KRAS mutations at a lower level, and whether such mutations had any bearing on the patients’ response to anti-EGFR therapy. Between 7% and 20% of CRC cases characterised as wild-type by Sanger sequencing or real-time PCR were found to harbour KRAS codon 12 or 13 mutations using pyrosequencing, Therascreen kits, locked nuclei acid PCR or mutant-enriched PCR techniques.19–22 However, the clinical data of these studies were conflicting as to whether the CRCs found to be KRAS mutant with more sensitive assays showed response to anti-EGFR therapy.19–22
Data regarding intratumour mutation heterogeneity are likely to evolve especially as more sensitive assays are developed. However, the following practical guidance can be issued at present. If both types of tissue are equally available, analysis of blocks from resected CRC is preferable to that of endoscopic biopsies. If only biopsy tissue is available and yields a wild-type genotype, there is currently insufficient evidence to require a repeat biopsy to exclude the possibility of a low-level mutation not being sampled in the first biopsy.
Rather than influencing patient stratification to receive anti-EGFR therapy at the outset, the presence of low-level RAS mutation (in an otherwise wild-type CRC) is more likely to be important in predicting for future resistance to anti-EGFR therapy. These mutant clones are thought to pre-exist in small numbers, but are encouraged to outgrow by anti-EGFR therapy and, when present in sufficient quantity, will manifest clinically as resistance to this therapy.23 ,24
Preanalytical aspects
How preanalytical preparation of tissue specimens affects subsequent molecular testing is reviewed elsewhere in great detail,25 ,26 and the following covers issues specifically related to RAS testing of CRC.
The majority of CRC tissue tested for RAS mutation comprises formalin-fixed biopsies or surgically resected primary tumours. In the latter case, there may be a delay in fixation of the specimen, or suboptimal fixation due to the large bowel not being opened and washed out, and/or due to dissection of a partially fixed specimen. Delayed or suboptimal fixation results in DNA degradation mainly through apoptosis or necrosis, whereas prolonged storage in formalin causes DNA degradation through excessive crosslinking.25 ,26 In both circumstances, the consequences of suboptimal DNA quality are a reduced sensitivity for detection of mutations and/or an increased failure rate. Formalin fixation can also lead to deamination of cytosine nucleotides, which can then result in detection of artefactual C>T/G>A mutations.26 ,27 While Bouin's fixative is no longer commonly used in the UK, testing of older tissue blocks may include tissue previously processed with this fixative. A warning sign is complete yellow discolouration of the tissue within the block and/or the eluent produced during DNA extraction of the tissue. In our experience, there is a higher failure rate of molecular testing from Bouin's fixed-tissue blocks. While this may partly be due to the age of the tissue prepared with Bouin's fixative, both the picric acid and acetic acid components of Bouin's fixative are recognised to accelerate degradation of nuclei acid.28 In some hospitals, endoscopic biopsies are mounted on acetate strips prior to fixation. Such strips can result in a poorer DNA yield if not excluded from tissue samples before DNA is extracted using a paramagnetic particle system, such as the Promega Maxwell 16 System. Provided neoplastic cell content has been recorded as suitable for molecular analysis (see below), there is little technical relevance whether tissue is cut onto sections or cut as curls straight for DNA extraction. However, it is recommended that both curls and sections undergo DNA extraction as soon as possible after cutting, to reduce the potential for DNA oxidation.
Neoplastic cell content
Accurate quantification of neoplastic cell content is crucial when investigating for somatic mutations in tissue which contains a mixture of neoplastic and non-neoplastic cells, for example, inflammatory and stromal cells (figure 1). The proportion of neoplastic DNA in a final extract is best reflected histologically by the proportion of neoplastic nuclei out of all nuclei in the tissue tested, rather than, for example, the proportion of area occupied by the whole neoplasm (figure 1). To this effect, and of particular relevance to CRC, areas of necrosis and acellular mucin (figure 1) should not be included in calculations of neoplastic cell content and should also be excluded from the macrodissection process where possible. The need for macrodissection of the tested tissue to enrich for neoplastic DNA should be governed by the limit of detection (LOD) of the assay used. It is acknowledged that aneuploidy (ie, multiple copies of one particular allele, whether mutated or not) is very common in most cancers, and can potentially result in increased (if mutant alleles are amplified or wild-type alleles are deleted) or decreased (if the wild-type allele is amplified) sensitivity of detection. However, as a general recommendation for this document and to account for heterozygous mutations, the minimum neoplastic cell content tested should be at least two times the assay's LOD. For example, tissue with at least a 10% neoplastic cell content is suitable for testing with an assay with a demonstrated LOD of 5%. There is varying interobserver reproducibility in the assessment of neoplastic cell content of CRC (as has been documented by the UK NEQAS Molecular Pathology KRAS EQA Scheme—ZC Deans, personal communication, 2014). Therefore, individual laboratories may wish to incorporate a safety margin and use a higher ratio of minimum neoplastic cell content to assay LOD. Further, this ratio is of less clinical relevance if a mutation has been equivocally demonstrated from the tested tissue.

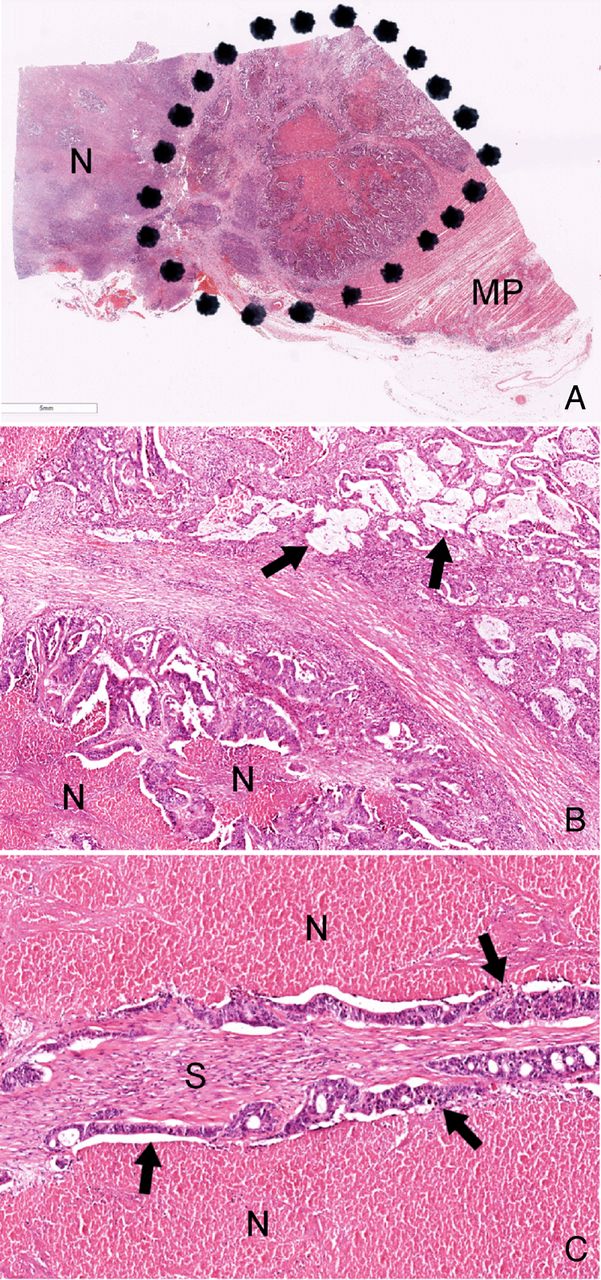{kind=link}
(A) Low-power image of a histological section taken from a colonic tumour tissue block submitted for RAS analysis. The tissue block includes a large area of necrosis (N) as well as background physiological tissue (eg, muscularis propria, MP). Therefore, only the area containing viable carcinoma (ie, within the black dots) should be macrodissected for DNA extraction. (B) Even within this area there are acellular mucin lakes (examples arrowed) and foci of necrosis (N); neither should be included in calculations of neoplastic cell content. (C) High-power field showing areas of necrosis (N) on either side of strips of adenocarcinoma epithelium (examples arrowed). Between these epithelial strips there is benign stroma which contains inflammatory cell and stromal cell nuclei. The neoplastic cell content of this particular high-power field has been calculated as an artificial but educational exercise for this guidance document. The areas of necrosis are ignored, but the stromal and inflammatory cell nuclei are included in the calculation of the field's neoplastic cell content, which is estimated as 60–70%.
Analytical aspects
Assay choice
A variety of assays have been used in the UK for KRAS testing. In our experience and in support of data from UK NEQAS Molecular Pathology,29 the KRAS assays that have been commonly used by NHS laboratories include in house pyrosequencing assays, COBAS, Therascreen pyrosequencing or RGQ kits, Sanger sequencing and HRM analysis. There have already been several comparison studies looking at the different performance characteristics of several of these KRAS assays.30–32 As newer technologies continue to be introduced, more of these comparison studies are anticipated. At the time of writing this document, technical aspects (including LODs and limitations) of the abovementioned assays had been collated in detail and analysed as part of the NICE Diagnostics Assessment Programme for KRAS testing of CRC; publication of the Programme's findings is pending. Some of the technical aspects of these assays have also been recently outlined elsewhere.33 However, table 1 represents a summary of the more important of these technical aspects.
Summary of test characteristics of the most commonly used assays in the UK for RAS testing of colorectal carcinoma to guide anti-EGFR therapy
Assay choice may be influenced by the more recent data on KRAS and NRAS which suggest that the presence of any RAS mutation (regardless of its codon location or specific nucleotide change) is sufficient to predict for resistance to anti-EGFR therapy.34–36 Therefore, there may be a future emphasis on rapid detection of a RAS mutation rather than on characterising its exact codon location and nucleotide change. However, these ‘screening’ approaches may limit the possibilities for subgroup analysis of mutations for research purposes or hypothesis generation.
Assay validation
Several aspects of a RAS assay require validation before the assay can be used for clinical cases.
The LOD of the assay should be calculated to determine the exact proportion of mutant alleles that can be detected by that assay. LOD can be accurately calculated using DNA extracted from isogenic combination of cell lines carrying known RAS mutations mixed with RAS wild-type cell lines to yield specific proportions of mutant allele. Alternatively, artificial mixes of DNA (whether genomic or plasmid) with precise quantification of mutant alleles can be used for this calculation.
The assay's precision and accuracy need to be analysed and recorded. In the context of detection of RAS mutations using qualitative assays, precision refers to how reproducibly the assay can detect the same mutation, whereas accuracy refers to whether or not the assay can detect reference genotypes, whether mutant or wild-type. Precision can therefore be assessed through repeat analysis of the same DNA sample within the same run, between runs and between operators at different times and in different conditions. Accuracy encompasses key aspects of a qualitative test (including its sensitivity and specificity), and is best assessed using clinical samples which have been genotyped either with a different, previously validated assay in the same laboratory or by the same assay in a different laboratory.
The number of clinical specimens required for validation depends on the statistical power required in each laboratory and for each test. For example, a validation performed with 100% experimental sensitivity (i.e., all results are correct) using only 30 specimens will lead to a statistical chance of a false negative of 10%, and therefore the sensitivity of the assay is predicted to be 90%.37 However, the same experimental sensitivity using 300 specimens predicts for a test sensitivity of 99%.37 Choosing the appropriate level of validation depends on the clinical setting and the intention of the test, though current literature lacks any firm indication of the optimal level of validation for RAS testing of CRC. A recent publication from the College of American Pathologists has suggested validation with at least 40 specimens,9 though it is noted that, based on the above formula, a perfect correlation with 40 specimens predicts for a test sensitivity of 92.5%.
When a CE-marked IVDD-compliant test is being used, a process of verification (as is outlined in further detail elsewhere37) rather than formal validation, is at least required to ensure that the test manufacturer's specifications are met in the laboratory which is starting to use the test.
The gold standard assay against which newer assays should be validated is controversial, and it is acknowledged that a combination of several assays could be used as one gold standard.37 However, it is recommended that a sequencing assay (Sanger or pyrosequencing) can act as that gold standard providing the neoplastic cell content is carefully chosen in relation to the LOD of the sequencing assay.
Test failure
It is our experience that most individual test failures during RAS testing of CRC are due to insufficient quantity and/or poor quality of the DNA template due to preanalytical issues, as is suggested by global failure across all the different KRAS and NRAS assays.
The quantity of neoplastic DNA template is affected by: the size of the specimen (with cytology specimens and whole tumour resections representing either end of the spectrum); the extent of macrodissection; and the proportion of neoplastic tissue remaining in the block received for molecular analysis (this proportion may be greatly depleted by preceding analyses such as multiple rounds of immunohistochemistry). If there is insufficient neoplastic tissue in the block received, further CRC tissue from the patient (such as another block from the resection specimen or another archived pathology specimen or, if these are not available, a repeat biopsy) should be requested. Paradoxically, too much DNA template can also cause test failure due to either the direct effects of excessive DNA or the presence of inhibitors coeluted with the DNA. In such instances, simple dilution of the DNA may allow a repeat test to succeed.
Quality of the DNA template is typically affected by fixative-related degradation as described above. This can lead to higher failure rates, particularly with assays that use large amplicons (i.e., >300 bp); redesigning primers to amplify shorter amplicons may result in better performance.
If only some of the KRAS or NRAS assays are failing, technical aspects of the failing assays need to be investigated. However, in the interim, the sample should be analysed with a back-up assay if available within the same laboratory, or sent to a second laboratory for immediate testing.
Reporting and interpretation
A minimum content of KRAS reports for guiding molecularly targeted therapy of CRC has already been recommended by UK NEQAS Molecular Pathology through its KRAS EQA scheme (ZC Deans, personal communication, 2014). Table 2 outlines a minimum content of reports for RAS testing of CRC, as is recommended by this guidance document.
Minimum content of reports for RAS testing of CRC to guide anti-EGFR therapy
Up until recently, it had been debated whether KRAS codon 13 mutations confer less resistance to anti-EGFR therapies.38 However, this controversy appears less relevant now with the more recent data derived from the FIRE-3, PEAK and PRIME clinical trials.34–36 The cumulative data of these trials indicate that, among CRC cases which are KRAS codon 12 and 13 wild-type, the presence of mutation in KRAS codons 59, 61, 117 or 146, or NRAS codons 12, 13, 59 or 61 will predict for poorer response to either cetuximab or panitumumab.34–36 The CRC cases studied in these trials showed no evidence of NRAS codon 117 or 146 mutations.34–36 Therefore, at the time of writing this document, it is recommended that RAS testing of CRC to guide anti-EGFR therapy should include analysis of at least KRAS codons 12, 13, 59, 61, 117 and 146 and NRAS codons 12, 13, 59 and 61.
Quality control and assurance
Internal
The validation of an assay's precision (see above) should provide the laboratory with an idea of whether duplicate testing is required. Most modern RAS assays show high precision, and their use by experienced laboratories would generally negate the need for duplicate testing in every case. For similar reasons, it may be argued that dual testing using two different assays is unnecessary, noting also that this would have an effect on accuracy rather than precision. However, an alternative assay for RAS is considered desirable as a back-up test for cases which fail or yield equivocal results with the first-line assay. A record should be kept of the proportion of test failures and, in each case, a likely reason for such failure. For every batch of samples analysed, a minimum of a positive control and a negative control (including a non-template control) per target analysed are recommended. For assays aiming to report low-level mutations, it is recommended that the LOD is analysed and recorded regularly by including known DNA samples with the required low level of mutant allele burden.
External
Any laboratory offering a RAS testing service for clinical cases must be involved in a RAS external quality assurance scheme, for example, those run by UK NEQAS Molecular Pathology, or the European Society of Pathology. Such a laboratory should also be appropriately accredited. In the UK, this is most commonly through the UK Assessment Service/Clinical Pathology Accreditation (UK) or, in the near future, through implementation of the ISO15189 standard. Finally, it is encouraged that laboratories participate in a sample exchange programme with other laboratories to allow for cross-analysis of, in particular, samples yielding failed or equivocal results.
Audit
Audit standards to which RAS testing laboratories should perform, are based on the data accumulated from previous RAS genotyping of CRC.34–36 Therefore, laboratories should expect approximately 40% of their tested CRC cases to show KRAS exon 2 mutations, approximately 6% to show KRAS exon 3/4 mutations, and approximately 3% to show NRAS exon 2/3 mutations. It should be noted that these proportions are indicative of the total population. Further studies are awaited to establish whether these proportions vary according to differing patient and tumour characteristics, such as tumour site; as an example of the latter with another gene, BRAF mutations are more commonly found in right-sided than left-sided CRCs.39
Turnaround time
As most RAS testing of metastatic CRC is being performed on-demand (certainly in the UK at the time of writing this document), a crucial aspect of this service is turnaround time (TAT). There are several potential definitions of TAT in this setting, that is, the time to issuing of a final RAS report from: (1) the clinical request for RAS testing, (2) the request of histological tissue from its source laboratory, or (3) the receipt of the tissue block at the testing laboratory. It is the last definition which is most commonly used, as is by guidelines which suggest that a KRAS codon 12 and 13 test result should be available within 7 working days.8 ,33 Using the same definition, a previous stipulation of commercial funding for such testing in the UK was a maximum TAT of 5 working days. This target, however, may now be more challenging in view of the greater number of KRAS codons and the addition of NRAS codons being analysed. Different laboratories may adopt different approaches to RAS multiplex testing with some implementing a sequential approach; for example, KRAS codons 12 and 13 (representing approximately 80% of RAS mutations in CRC)34–36 are analysed first and if these are wild-type, the remaining codons are studied. An alternative approach is blanket testing of all the RAS codons. However, even here, there has been variation in how this is performed with some laboratories choosing a screening assay (e.g., single-strand conformation analysis or HRM) and then a sequencing assay for the codon found to bear mutation. From a patient perspective, when results are required for immediate therapeutic decision making, it is not acceptable that an increased range of biomarker testing leads to ever increasing TAT and potential treatment delays. Therefore, it is recommended that RAS testing (using the above recommended panel) should be completed and reported in >90% of specimens with a TAT of ≤7 working days from receipt of the specimen in the testing laboratory.
Future developments
There are increasing reports of RAS wild-type CRC patients who first show response to EGFR inhibitors, but who later demonstrate RAS mutations with progressive disease, suggesting acquired resistance to these drugs.23 ,24 There is a great need to characterise how frequently, widely and quickly these resistance-mediating mutations arise. Recent data demonstrate that KRAS mutations may be characterised with very high sensitivity and specificity from circulating tumour-derived, cell-free DNA.40 Analysis of such ‘liquid biopsies’ may have a future clinical role particularly for detecting emerging RAS mutant clones which mediate resistance to anti-EGFR therapy.
In the same way that additional mutations in KRAS and NRAS have now been incorporated to further triage patients for anti-EGFR therapy, it is likely that similar roles for other genes will be demonstrated in the future. At the moment, it remains controversial whether BRAF represents one such gene, although there is strong evidence that BRAF mutant metastatic CRC has a poor prognosis irrespective of treatment.34 Nonetheless, with the introduction of novel treatment strategies, more corresponding biomarkers will be needed. In this sense, incorporation of BRAF mutation analysis for metastatic CRC may become essential if current or future clinical studies assessing combinations of BRAF and EGFR inhibitors are successful. As the gene panel of predicting anti-EGFR therapy response expands and new therapies become available, laboratories will need to rapidly respond in extending their multiplex testing of CRC. Next-generation sequencing would seem an ideal platform for such multiplex testing.
Conclusion
RAS genotyping of CRC to guide anti-EGFR therapy is ever evolving and rapidly being updated. At the current time, the main recommendations for RAS testing of CRC, at least in the UK, are presented in box 1. It is anticipated that this document will be updated, particularly if a quantum leap in such testing occurs in the future.
Acknowledgments
The authors thank Dr ZC Deans for contributing data from UK NEQAS Molecular Pathology.
References
Footnotes
-
NACSW, DG, MS-T are lead authors.
-
Contributors All the authors contributed to the idea, conception and contents of the document. NACSW, DG and MST wrote the first drafts of the document; the remaining authors reviewed and/or amended these drafts.
-
Competing interests DG has received honoraria from Roche Molecular Systems, Roche Molecular Diagnostics, Roche Products Ltd and Amgen, and received grant support from Roche Molecular Systems. MS-T is a regular member of advisory boards for Roche Molecular and Ventana Roche, and has been invited to speak in events organised by these companies. He is part of a UK Technology Strategy Board grant for product development (STRATFix) coordinated by Qiagen. ES works for Cancer Research UK on the Stratified Medicine Programme which is jointly funded by AstraZeneca and Pfizer, and is also working with Qiagen on a project investigating novel tissue fixatives and preservatives. NACSW has received consumables support for research from Ventana Roche.
-
Provenance and peer review Not commissioned; internally peer reviewed.