Article Text

Abstract
Aims Megakaryocyte expansion in myeloproliferative neoplasms (MPNs) is due to uncontrolled proliferation accompanied by dysregulation of proapoptotic and antiapoptotic mechanisms. Here we have investigated the intrinsic and extrinsic apoptotic pathways of megakaryocytes in human MPNs to further define the mechanisms involved.
Methods The megakaryocytic expression of proapoptotic caspase-8, caspase-9, Diablo, p53 and antiapoptotic survivin proteins was investigated in bone marrow specimens of the MPNs (n=145) and controls (n=15) using immunohistochemistry. The megakaryocyte percentage positivity was assessed by light microscopy and correlated with the MPN entity, JAK2V617F/CALR mutation status and platelet count.
Results The proportion of megakaryocytes in the MPNs expressing caspase-8, caspase-9, Diablo, survivin and p53 was significantly greater than controls. A greater proportion of myeloproliferative megakaryocytes expressed survivin relative to its reciprocal inhibitor, Diablo. Differences were seen between myelofibrosis, polycythaemia vera and essential thrombocythaemia for caspase-9 and p53. CALR-mutated cases had greater megakaryocyte p53 positivity compared to those with the JAK2V617F mutation. Proapoptotic caspase-9 expression showed a positive correlation with platelet count, which was most marked in myelofibrosis and CALR-mutated cases.
Conclusions Disruptions targeting the intrinsic apoptotic cascade promote megakaryocyte hyperplasia and thrombocytosis in the MPNs. There is progressive dysfunction of apoptosis as evidenced by the marked reduction in proapoptotic caspase-9 and accumulation of p53 in myelofibrosis. The dysfunction of caspase-9, which is necessary for proplatelet formation, may be the mechanism for the excess thrombocytosis associated with CALR mutations. Survivin seems to be the key protein mediating the megakaryocyte survival signature in the MPNs and is a potential therapeutic target.
- MYELOPROLIFERATIVE DISEASE
- APOPTOSIS
- IMMUNOHISTOCHEMISTRY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Myeloproliferative neoplasms (MPNs) are a group of clonal proliferative bone marrow diseases characterised by somatic mutations (eg, JAK2V617F, CALR frameshift lesions)1–5 and varying hyperplasia of the myeloid lineages. Megakaryocyte hyperplasia with clustering and associated morphological atypia with pleiomorphism are key diagnostic histological features.6–8 The pathobiological basis underlying these numerical and morphological megakaryocytic abnormalities is thought to result from multiple molecular disruptions promoting proliferation and enhancing survival.9–13 These megakaryocytes have impaired death mechanisms conferred by overexpression of antiapoptotic Bcl-XL and reductions in pro-death BNIP-3.9 ,11–13 These changes are universal in the MPNs but there are differences between entities. Megakaryocytes in essential thrombocythaemia (ET) have been shown to have a more proliferative profile, whereas in myelofibrosis (MF) they exhibit greater proapoptotic impairments.9 ,13 These changes occur irrespective of the JAK2V617F or calreticulin (CALR) driver mutations, although those with a CALR lesion have greater proapoptotic dysfunction.13 The mechanisms driving this apoptotic dysregulation in megakaryocytes in the MPNs have not been explored.
Apoptosis is mediated via extrinsic and intrinsic apoptotic cascades (figure 1), with both pathways being capable of inducing programmed cell death following exposure to apoptotic insults and the accumulation of excess DNA damage.14–16 These pathways converge at the cleavage of procaspase-3 to produce active caspase-3, which is capable of committing the cell to apoptosis.14–16 Alterations in caspase biology, including caspase-8 (extrinsic) and caspase-9 (intrinsic), have been implicated in a number of malignancies in humans and animal models. In caspase-8-deficient mice, B-lymphocytes have impaired cytokinesis and chromosomal instability, and show a tendency towards lymphoma development.17 Similarly, caspase-9 gene polymorphisms and its downregulation are associated with solid tumours and their malignant progression.18–20 Both caspase-8 and caspase-9 are potentially important in regulating megakaryocyte turnover in the MPNs. Frameshift lesions targeting CALR may disrupt megakaryocyte apoptosis through its inability to facilitate caspase-8 activation and antiapoptotic protein cleavage.21 While the precise pathogenesis surrounding CALR lesions and megakaryocyte apoptosis is unknown, its mutated product is exclusively expressed in myeloproliferative megakaryocytes.22 ,23 In megakaryocytes, caspase-9 activity appears necessary for proplatelet formation.24 Some reports suggest that caspase-9 may be redundant, with CASP9 deletions in ex vivo-cultured mouse megakaryocytes impairing procaspase-3 activation, and encouraging both megakaryopoiesis and proplatelet formation.25 This does not however discount the presence of other caspases to conduct cytoskeletal proplatelet fragmentation in the absence of caspase-9. Moreover, caspase-9 loss may enhance the tumourigenic potential of megakaryocytes as their proliferative capacity increases.25

Diagrammatic representation of the extrinsic and intrinsic apoptotic pathways. The extrinsic (receptor-mediated) apoptotic pathway commences following FasL binding the Fas death receptor. The union forms a death complex that is capable of recruiting FADD and procaspase-8 to form the DISC. The DISC recruits procaspase-8 proteins that undergo reciprocal cross-proteolysis and proximity-induced dimerisation to form an active caspase-8 that targets procaspase-3. The intrinsic (mitochondrial-mediated) apoptotic pathway relies on the insertion of homo-oligomerised pro-death Bcl-2-family proteins (Bak and Bax) into the outer mitochondrial membrane. The release of cytochrome-c subsequently ensues and acts to recruit procaspase-9 and Apaf-1 to form the heptameric apoptosome. The apoptosome recruits procaspase-9 molecules, which reciprocally cleave one another to form an active caspase-9 that targets procaspase-3. Both caspase-8 and caspase-9 then cleave procaspase-3 to form the executioner caspase-3, which brings about coordinated cell death. Survivin aims to inhibit caspase-9 activation while simultaneously competing with the inhibitory action of cytosolic p53 and Diablo. Nuclear p53 also activates the transcription of pro-death molecules required in both apoptotic cascades. Apaf-1, apoptotic protease activating factor-1; Bak, Bcl-2 homologous antagonist/killer; Bax, Bcl-2-associated X protein; Casp-3, caspase-3; Casp-8, caspase-8; Casp-9, caspase-9; Cyt-c, cytochrome-c; DISC, death-inducing signalling complex; FADD, Fas-associated protein with death domain; Fas, Fas death receptor; FasL, Fas receptor ligand; Procasp-3, procaspase-3; Procasp-8, procaspase-8; Procasp-9, procaspase-9.
Apoptotic cell death via the intrinsic pathway is regulated by inhibitors of apoptosis proteins (IAP) that specifically constrain the pro-death actions of caspase-9. One of these IAP, survivin, restricts the IAP-inhibitor Diablo protein and prevents it from activating caspase-9.26–29 This impedes the intrinsic apoptotic pathway and confers a cytoprotective effect. The relevance of Diablo has been shown in human tumours: downregulation has been associated with progressive disease and poor survival in both solid and haematological malignancies.30–33 Survivin overexpression is well documented in leukaemias34–37 and lymphomas38–41 where it increases the survival capacity of affected tumour cells. On the other hand, survivin loss causes mitotic catastrophe characterised by cell death or polyploidisation.37 ,42 Mouse megakaryocytes failing to express survivin have limited proliferation but increased ploidy, with loss of the IAP preferentially selecting for those megakaryocytes capable of escaping intrinsic apoptotic fates.42 Megakaryocytes are known to express survivin and during anaphase it restricts cytokinesis to enable polyploidisation.43 The relationship between survivin, Diablo and caspase-9 is complex and has not been assessed in megakaryocyte survival in the MPNs.
p53, the ‘master’ regulator of cell cycle, is also involved in the apoptotic machinery. Its regulatory role involves arresting the cell cycle, initiating senescence and inducing DNA repair mechanisms.44 ,45 Failing these, p53 activates the intrinsic apoptotic cascade by binding Bak/Bax to induce cytochrome-c release while simultaneously inhibiting antiapoptotic Bcl-2-related proteins46 ,47 and survivin.48 The role of p53 in megakaryocytes is disputed. Ex vivo-cultured megakaryocytes have been shown to express low levels of p5349 and its absence in p53-null mice has no effect on megakaryopoiesis.50 However, the in vitro knockdown of p53 has been reported to increase megakaryocyte proliferation and regulate both its ploidy and differentiation.51–54 This disparity between in vitro and ex vivo experimental settings ultimately suggests that in vivo, p53 loss is tolerated in the presence of other compensatory proapoptotic mechanisms. In the MPNs, alterations in p53 have not been linked to megakaryocyte hyperplasia although mutations targeting TP53 do occur during their leukaemic transformation.55–57
Apoptotic signalling is crucial to megakaryocyte polyploidisation and platelet production. Limited studies have identified that apoptotic signalling processes are disrupted in megakaryocytes of the MPNs.9–13 We intended to further delineate the biological basis of the apoptotic disturbances affecting megakaryocytes in the MPNs by assessing several biomarkers implicated in the intrinsic and extrinsic apoptotic pathways. We demonstrate through immunohistochemical analyses of human MPNs that the enhanced survival of myeloproliferative megakaryocytes occurs through inhibition of intrinsic death effectors despite concurrent extrinsic apoptotic activation.
Materials and methods
Patient samples
Bone marrow trephine (BMT) specimens from patients with polycythaemia vera (PV), ET and MF (including both primary MF and post-PV and post-ET MF) (n=145) and normal bone marrow controls (n=15) were collected from patients through PathWest Laboratory Medicine (Western Australia, Australia) and Queen Mary Hospital (Hong Kong SAR, China) between 1999 and 2015. Of the 145 patients with MPNs, 133 were untreated at the time of bone marrow collection. The PathWest specimens were formalin-mercury fixed while those from Hong Kong were fixed in formalin. All BMT specimens were acid decalcified and paraffin embedded. Morphological review of all cases was undertaken in accordance with WHO criteria7 and classified according to MPN entity (table 1). PathWest BMT specimens were processed using a TMA Master tissue microarrayer (3DHistech, Australia) to create tissue microarrays (TMA) as outlined in Malherbe et al.13 Whole BMT sections from the Hong Kong cohort were used for immunohistochemical investigation. JAK2V617F mutation testing was performed by allele-specific PCR analysis. CALR mutations were detected using methods outlined by Nangalia et al.5 MPL mutation testing was not performed and cases negative for JAK2V617F and CALR mutations were classified as ‘double negative’ (DN). Platelet counts (×109/L) were recorded for each MPN case at the time of collection of the BMT specimen.
Summary of the cohort studied
Immunohistochemical staining
Whole BMT specimens and TMA were sectioned at 4 μm onto charged glass slides (Hurst Scientific, Australia). Monoclonal antibodies were to formalin-mercury-resistant epitopes and were validated on control tissue prior to their application on BMT sections. Antibodies used were to CD61 (clone 2f2, Leica Biosystems, Australia), caspase-8 (clone 90A992, Thermo Scientific Pierce Antibodies, Australia), caspase-9 (clone F-7, Santa Cruz Biotechnology, USA), p53 (clone DO-7, Leica Biosystems, Australia), survivin (clone 71G4B7, Cell Signaling Technologies, USA) and Diablo/Smac (clone D5S3R, Cell Signaling Technologies, USA). All immunohistochemical staining was performed on an automated Leica BOND RX immunostainer (Leica Biosystems) as outlined by Malherbe et al.13 Positive and negative megakaryocytes were counted by a minimum of two observers and the percent positive calculated for each apoptotic biomarker. Megakaryocyte-rich (≥50) areas within whole BMT specimens were selected for enumeration of each biomarker up to a maximum of 200 megakaryocytes. Observers were blinded to both the diagnostic entity and mutation status of cases. Tissue areas and/or megakaryocytes of interest were photographed using a Pixera Pro 600ES microscope camera (Pixera, USA).
Statistical analysis
Mean megakaryocyte percentage positivity and SD were calculated according to the MPN subtype and JAK2V617F/CALR mutational status for each apoptotic biomarker. Significant differences between the MPNs and control megakaryocyte positivity were assessed using Mann–Whitney U tests. One-way Kruskal–Wallis analysis of variance (ANOVA) analyses with post hoc Dunn's tests were performed to evaluate megakaryocyte positivity variations in relation to subtype and JAK2V617F/CALR mutation status. Platelet counts for the MPNs stratified according to subtype and JAK2V617F/CALR mutation status were correlated with the mean megakaryocyte expression for all biomarkers using Spearman's correlation. Significant differences were set at p<0.05. All statistical analyses were conducted using GraphPad Prism V.6 software (GraphPad Software, USA).
Results
MPNs versus controls
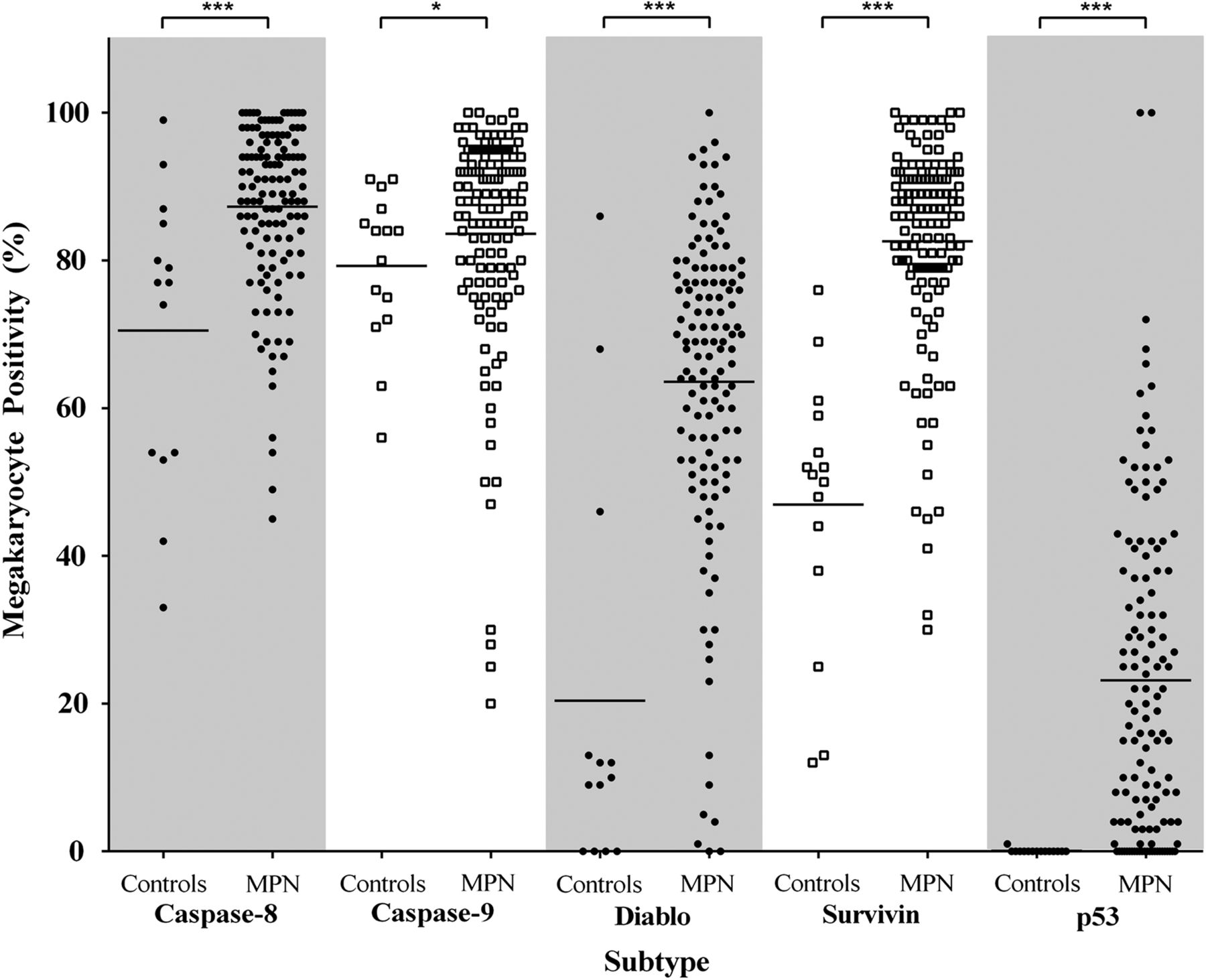
Megakaryocytes were visually identified on morphology, antigen expression (red chromogen) and nuclear haematoxylin counterstain. All antibodies produced the expected expression profile; there was no non-specific background staining in any sections. CD61 was used to confirm the identity of megakaryocytes in BMT with marked atypia (eg, MF). Mean megakaryocyte positivity was significantly greater in the MPNs than in controls for all apoptotic biomarkers, that is, caspase-8, caspase-9, survivin, Diablo and p53 (figure 2). Caspase-8 showed significantly more (∼16.8%) positive megakaryocytes in the MPNs than controls, p=0.0005. A similar significant trend was observed for caspase-9, although this increase was smaller (∼4.3%), p=0.023. Of note was that megakaryocytes present within clusters, a key feature of the MPNs, showed strong positivity for both caspase-8 and caspase-9, whereas single intertrabecular or paratrabecular megakaryocytes were more commonly negative (figure 3A–C). There were significantly greater numbers of Diablo and survivin-positive megakaryocytes in the MPNs than controls, ∼3.1-fold, p<0.0001 and ∼1.8-fold, p<0.0001, respectively. When comparing the proportion of Diablo and survivin-positive megakaryocytes in the MPNs, the balance was in favour of survivin by ∼19.0%. The cytoplasmic expression of Diablo was weak and heterogeneous among megakaryocyte clusters in the MPNs (figure 3D). Contrastingly, myeloproliferative megakaryocytes showed strong nuclear localisation of survivin (figure 3E, F). p53 was positive (nuclear) in myeloproliferative megakaryocytes (∼23.1%) and virtually absent from controls, p<0.0001 (figure 3G, H).

Percentage of positive megakaryocytes in myeloproliferative neoplasms (MPNs) and controls for caspase-8, caspase-9, Diablo, survivin and p53. Mean megakaryocyte positivity in MPN cases was significantly greater for all biomarkers in comparison to controls. Statistically significant difference, p<0.05 (*), p<0.001 (***).
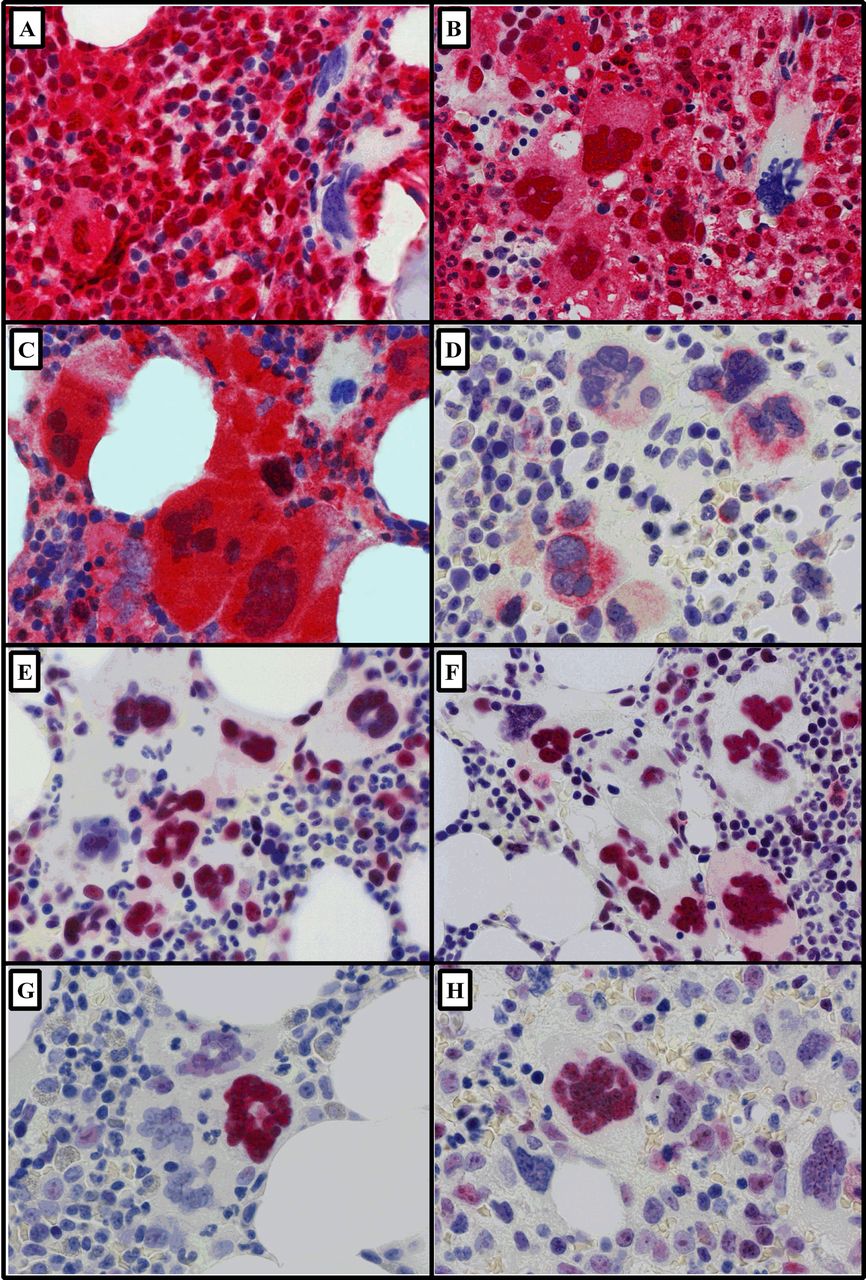
Representative images (×600) of immunohistochemically stained megakaryocytes in myeloproliferative neoplasms (bone marrow trephine sections; Fast Red chromogen and haematoxylin counterstain). (A) Caspase-8 in polycythaemia vera and (B) myelofibrosis (MF) and (C) caspase-9 in MF. (D) Diablo-positive megakaryocytes in essential thrombocythaemia (ET). Clusters of megakaryocytes showing strong positive nuclear expression for (E) survivin in ET and (F) in MF. In contrast, nuclear p53 was limited to isolated positive megakaryocytes among a negative majority (G) in ET and (H) in MF.
MPNs by disease entity and genotype
Since myeloproliferative megakaryocytes showed increased expression of apoptosis-associated antigens, we then proceeded to assess reactivity by morphological entity (ie, PV, ET and MF) and mutation status (ie, JAK2V617F+, CALRMut and DN: JAK2V617F−/CALRWT). Using Kruskal–Wallis ANOVA analyses, significant differences were seen between the MPN subtypes for caspase-9 and p53, p=0.0015 and p=0.0079, respectively (figure 4). Post hoc analyses showed caspase-9 expression to be significantly lower in MF megakaryocytes (∼77.9%) than both PV (∼90.3%, p=0.0032) and ET (∼85.6%, p=0.019) with a similar, borderline insignificant trend for survivin, Kruskal–Wallis ANOVA p=0.073. The number of p53-expressing megakaryocytes in MF was ∼2.7-fold greater than in PV trephines, p=0.0071. The differences in megakaryocyte positivity between the MPN entities for caspase-8 and Diablo were not significant, p>0.05 (data not shown).
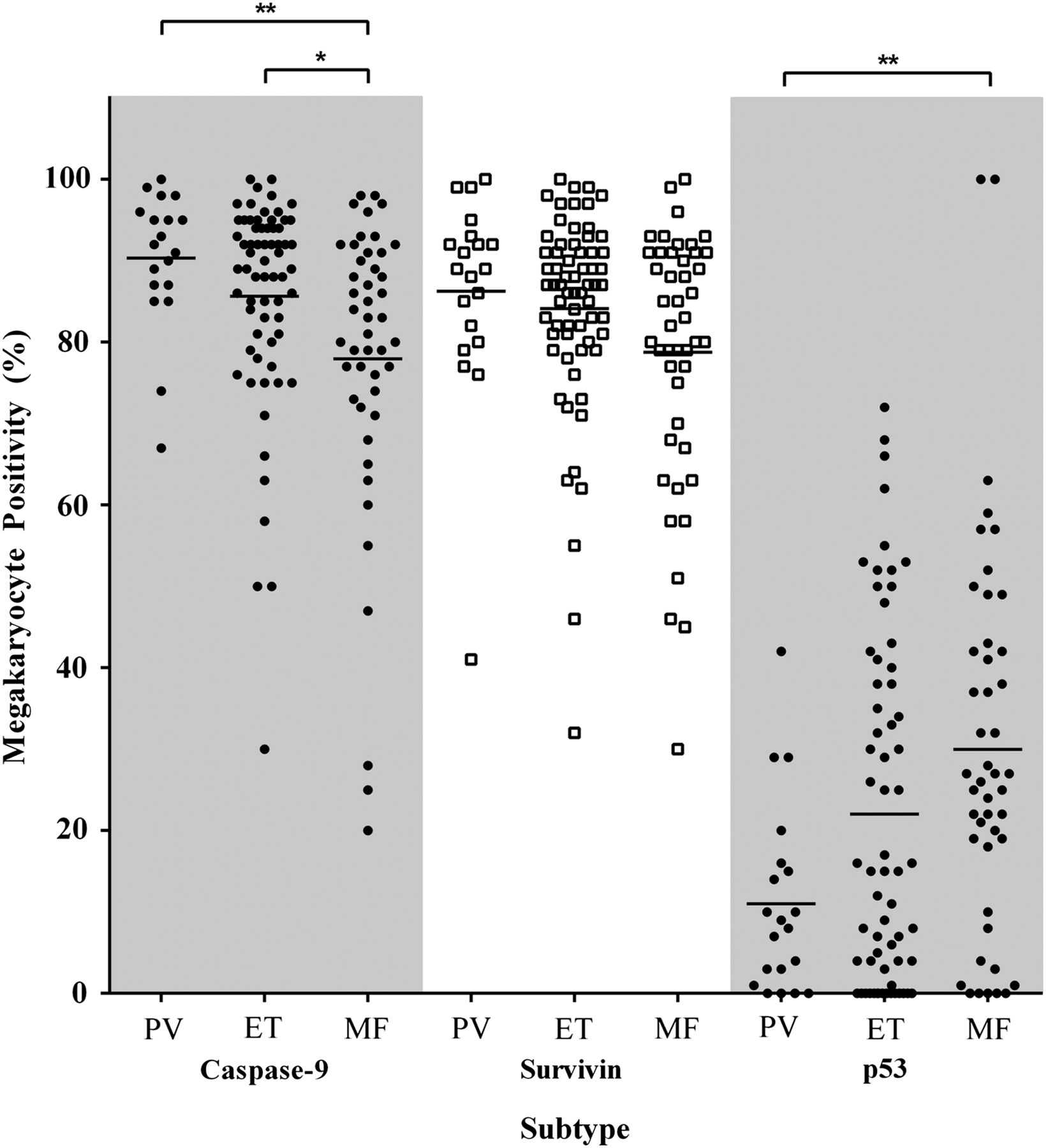
Percentage of megakaryocytes positive according to myeloproliferative neoplasm entity for caspase-9, survivin and p53. Mean megakaryocyte positivity for caspase-9 was significantly lower in myelofibrosis (MF) than polycythaemia vera (PV) and essential thrombocythaemia (ET), p=0.0032 and p=0.019, respectively. The per cent positive megakaryocytes in MF was lower for survivin (p=0.073) and higher for p53 than PV and ET, p=0.0071 and p=0.18, respectively. Statistically significant difference, p<0.05 (*), p<0.01 (**).
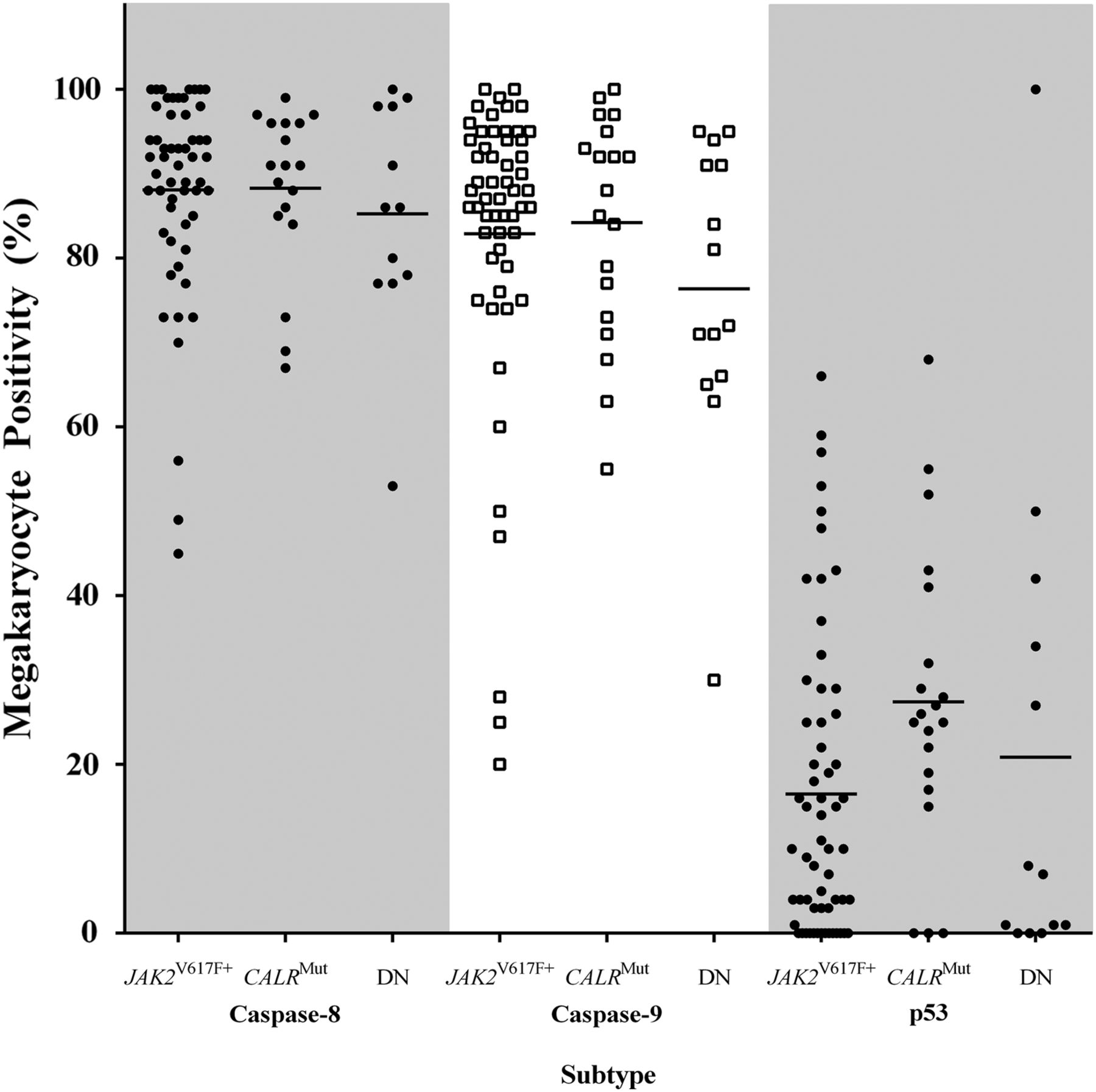
When apoptotic biomarkers were assessed by mutation status, no significant differences were seen, p>0.05. The proportion of p53-expressing megakaryocytes was greater in CALRMut than JAK2V617F+ trephines, although this difference was borderline insignificant, Kruskal–Wallis ANOVA p=0.0660, post hoc p=0.0667 (figure 5). Caspase-8-positive and caspase-9-positive megakaryocytes in JAK2V617F+ and CALRMut cases were similar and marginally increased when compared with JAK2V617F−/CALRWT (figure 5). Mean Diablo and survivin megakaryocyte positivity did not differ by genotype (data not shown).

Caspase-8, caspase-9 and p53 in myeloproliferative neoplasms by JAK2V617F and CALR mutation status. The percentage of caspase-8-positive and caspase-9-positive megakaryocytes were similar for JAK2V617F+ and CALRMut and not significantly different in comparison to JAK2V617F−/CALRWT cases, p>0.05. p53 megakaryocyte positivity was greater in CALRMut than JAK2V617F+, p=0.067. DN—double negative (ie, JAK2V167F−/CALRWT).
Apoptotic markers and platelet count
Since proplatelet production requires megakaryocyte apoptotic signalling,24 ,25 platelet counts were correlated with megakaryocyte positivity for all apoptotic biomarkers in the MPNs. There was a significant, weak positive correlation between caspase-9 expression and platelet count for all MPNs, r=0.28, p=0.0018 (figure 6A). This correlation was strongest for cases of MF, r=0.34, p=0.026 (figure 6B) and most notably those MPNs with CALRMut, r=0.50, p=0.030 (figure 6C). When cases that had received prior therapy were excluded from this analysis, these correlations remained significant (data not shown). No significant correlations existed between any of the other apoptotic biomarkers and platelet count, p>0.05 (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Caspase-9 megakaryocyte positivity correlated significantly with platelet counts for (A) all myeloproliferative neoplasm cases (r=0.28, p=0.0018), (B) myelofibrotic entities (r=0.34, p=0.026) and (C) CALR-mutated trephines (r=0.50, p=0.030).
Discussion
Previous studies have indicated that there is dysfunction of apoptotic mediators in the MPNs, that is, increased antiapoptotic Bcl-XL and reduced pro-death BNIP-3.9 ,13 However, the precise apoptotic disruptions promoting abnormal megakaryocyte accumulation and thrombocytosis remain unclear. Here we provide insight into these mechanisms and demonstrate that the enhanced survival of megakaryocytes in the MPNs may be a result of dysregulation of the intrinsic apoptotic pathway. The key protein mediating this survival signature appears to be survivin, an inhibitor of the intrinsic apoptotic cascade.27–29 While survivin impedes megakaryocyte death, the reciprocal increase of Diablo, its inhibitor,26 is insufficient and unlikely to nullify its antiapoptotic effect. Further, there is low-level upregulation of caspase-9, the main effector of the intrinsic apoptotic cascade (figure 1),16 and its expression correlated with platelet count. These data suggest that these aberrations facilitate megakaryocyte hyperplasia and thrombocytosis rather than directing apoptotic execution. The overexpression of pro-death caspase-8 (extrinsic) and p53 may be an attempt to counteract these changes. The progressive dysfunction of megakaryocyte apoptosis, which has previously shown to be most discernible in MF and CALR-mutated MPNs,9 ,13 can now be further supported by marked reductions in pro-death caspase-9 and accumulation of p53.
Survival advantages gained through inhibition of the intrinsic apoptotic cascade is a key pathological mechanism promoting megakaryocyte hyperplasia in the MPNs. We have previously demonstrated that myeloproliferative megakaryocytes show disrupted expression of the Bcl-2 family of apoptotic biomarkers (ie, upregulated antiapoptotic Bcl-XL, diminished pro-death BNIP-3) that regulate intrinsic apoptosis.13 We further support this theory by showing that survivin is strongly overexpressed in myeloproliferative megakaryocytes. Survivin acts to limit procaspase-9 cleavage, thereby constraining caspase-9 activation in megakaryocytes and impeding their death (figure 1).27–29 Furthermore, survivin assists megakaryocyte polyploidisation and its increase in the MPNs may be an obligatory survival prerequisite as the majority of these megakaryocytes are of high ploidy (up to 512N).43 ,58 Myeloproliferative megakaryocytes attempt to neutralise this survival advantage by expressing Diablo, which directly competes with the antideath effects conferred by excess survivin.26 However, a greater proportion of megakaryocytes in both controls (∼26.5%) and the MPNs (∼19.0%) expressed survivin than those with upregulated Diablo content. Further, the weak expression intensity of Diablo-positive megakaryocytes in the MPNs correlates with lower concentrations of the IAP-inhibitor in comparison to stronger survivin signals. This was not seen in control megakaryocytes, where the staining intensity of survivin and Diablo was similar. The survival advantage therefore persists in myeloproliferative megakaryocytes, and in conjunction with other intrinsic apoptotic disturbances is likely to promote their accumulation.
Our data indicate that myeloproliferative megakaryocytes attempt to counteract these survival effects by stimulating the extrinsic apoptotic cascade via caspase-8. This may represent a protective, death-promoting response against megakaryocyte oncogenesis following the accumulation of excess molecular abnormalities. Myeloproliferative megakaryocytes also increase their nuclear p53 content relative to controls where p53 was, and has previously been shown to be, mostly absent.49 We postulate that caspase-8 overactivity induces TP53 gene transcription to produce p53.44 ,45 p53 then translocates to the megakaryocyte nucleus where it activates the transcription of pro-death biomarkers (eg, Fas, Apaf-1) to stimulate both extrinsic and intrinsic apoptotic cascades.59 Moreover, megakaryocytic p53 is potentially increased to directly abrogate the antideath effects of survivin.48 Alternatively, this p53 upregulation in myeloproliferative megakaryocytes may be to regulate their polyploidisation by inducing states of megakaryocyte senescence.60
There were differences in the megakaryocyte apoptotic profiles according to mutation status. Most notably, CALR-mutated cases showed greater megakaryocytic p53, but not caspase-8 positivity, than those with the JAK2V617F mutation, although the former was of borderline insignificance. It has previously been shown that calreticulin is involved in caspase-8 activation.21 Our data suggest that the mutated calreticulin gene product does not influence caspase-8 levels. Rather, it appears that CALR lesions disrupt alternative apoptotic effectors and that affected megakaryocytes will attempt a remedial pro-death response dominated by p53 overexpression. We also show that the small increase in the proportion of caspase-9-positive megakaryocytes in the MPNs is positively correlated with platelet count, and that this correlation strengthens with CALR mutations. We propose two explanations for this data. First, it is likely that CALR lesions interfere indirectly with caspase-9, rather than with caspase-8. Second, the minor increase in caspase-9 is unlikely to enable megakaryocyte apoptosis to be completed via the intrinsic apoptotic pathway. Rather, several groups have shown that constitutively activated caspase-9 in the presence of intrinsic antiapoptotic effectors is necessary for proplatelet synthesis and shedding.24 ,61–63 Therefore, low-level caspase-9 upregulation and concurrent survivin overexpression, especially among CALR-mutated MPNs, may drive thrombocytosis instead of facilitating megakaryocyte apoptosis.
As MPNs progress towards MF, megakaryocytes accumulate additional morphological, topographical and molecular abnormalities, and show the greatest survival signature.7 ,13 ,64 The megakaryocytes in MF also have greater apoptotic disturbances than the other MPNs. This includes lower caspase-9 and greater numbers of p53-positive megakaryocytes than both PV and ET. It appears that the megakaryocyte attempts to rectify this apoptotic disruption by reducing survivin and overactivating the ‘p53-mediated’ transcription of its pro-death effectors required for apoptotic execution.59 This is consistent with the morphological appearances of megakaryocytes in MF with their greater nuclear pleiomorphism and para-apoptotic changes.7 ,13 ,65 Further, the reduction in number of caspase-9-positive megakaryocytes in MF may be the mechanism for the lower platelet counts characteristic of this phenotype compared with other MPN entities.7 ,66
In summary, disruptions targeting the intrinsic apoptotic cascade appear to promote megakaryocyte accumulation and thrombocytosis in the MPNs. Survivin seems to be a key mediator of this antiapoptotic signature and therefore could be a potential therapeutic target. Anti-survivin agents have been developed, which show therapeutic efficacy for many other malignancies.67–69 As such, targeting survivin in the MPNs could potentially control megakaryocyte accumulation and downstream effects (eg, stromal accumulation). Further, a synergistic option could potentially be achieved by including Diablo/Smac-like mimetics70–72 to augment innate and already upregulated megakaryocytic Diablo in the MPNs.
Take home messages
Dysregulation of intrinsic apoptotic mechanisms in megakaryocytes in the myeloproliferative neoplasms (MPNs) promotes their hyperplasia and thrombocytosis.
Survivin, a protein that inhibits the intrinsic pathway of apoptosis, appears to be a key mediator of apoptotic failure of megakaryocytes in the MPNs.
Caspase-9 dysfunction may explain the heightened thrombocytosis in MPNs, especially among those with CALR mutations.
Acknowledgments
JAJM received financial support from the Royal College of Pathologists of Australasia through the RCPA Scholarship in Pathology for Medical Schools 2015 for this study.
References
Footnotes
Handling editor Mary Frances McMullin
Contributors JAJM, KAF and BM performed the immunohistochemical studies. C-CS, H-WI and BBG conducted the molecular analyses. JAJM, KAF, SK, BM and WNE performed the megakaryocyte enumeration for all cases. JAJM conducted the statistical analyses. C-CS, HWI, CF and WNE undertook the morphological review. RH and WNE initiated the study. All authors contributed to the writing of the manuscript.
Funding The study was supported by the Australian Leukaemia and Lymphoma Group and Novartis Pharmaceuticals Australia Pty Ltd, the Cancer Council Western Australia, Perpetual Foundation-Ann Helene Toakley Charitable Endowment and the Royal College of Pathologists of Australasia.
Competing interests None declared.
Patient consent Obtained.
Ethics approval (1) Sir Charles Gairdner Hospital Human Research Ethics Committee (#2012-094). (2) The University of Western Australia Human Research Ethics Committee (#RA/4/1/6566). (3) The Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (#UW 13-189).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Data are available upon request and at the discretion of the authors.