Article Text

Abstract
SMARCB1 is the core subunit of the SWI/sucrose non-fermenting ATP-dependent chromatin remodelling complex located on the long arm of chromosome 22 (22q11.2). Since discovering genetic alterations of the SMARCB1 gene in malignant rhabdoid tumours, the family of tumours harbouring loss of SMARCB1 expression has been steadily expanding. In this review, we give a general overview of SMARCB1, its role in various cancers including germline mutations, association with genetic syndromes and role in future targeted therapies.
- MALIGNANT TUMOURS
- CARCINOMA
- GENETICS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
SMARCB1 is a ubiquitously expressed nuclear protein. The SMARCB1 gene is a core subunit of the SWI/sucrose non-fermenting (SNF) ATP-dependent chromatin remodelling complex, involved in the activation and repression of gene transcription.1–6
History of nomenclature
Since its discovery, this gene has been known by a variety of monikers. First, following its recognition in yeast, the gene was referred to as SNF5 (‘sucrose non-fermenting’) genes, in reference to its role in sucrose metabolism.3 ,7 ,8 The human homologue of the gene was later discovered from its interaction with the HIV integrase and was subsequently designated as integrase interactor 1,9 by which the corresponding immunohistochemical marker is now universally referred to as. Later, the gene was given an official Human Genome Organisation name of SWI (mating-type switching)/SNF-related matrix-associated, actin-dependent regulator of chromatin, subfamily B, member 1 (SMARCB1) and has now been incorporated into the cancer genome sequencing literature. In addition, an alternative appellation that has also gained momentum is BAF47. This refers to the BAF (Brahma (gene) (BRM) or BRG1 associated factors) subunit of the gene within the SWI/SNF complex and followed by the mass of the protein measured in kilodaltons, which is 47 kDa.
Structure
The SMARCB1 gene maps to the long arm of chromosome 22 at position 11.23 (22q11.23).10 The peptide sequence of the gene is encoded by nine exons (figure 1) and extends over ∼50 kb.10 SMARCB1 gene encodes one core subunit of SWI/SNF complex.3 ,4 ,6 ,11–13 These subunits of the SWI/SNF complex were discovered using yeast through two genetic screens to identify genes that control: (1) induction of Homothallic switching endonuclease (HO) endonuclease that is required for mating-type switching (swi) and (2) induction of the suc2 gene that encodes invertase enzyme (sucrose non-fermenting or snf). Each subunit contains a single ATPase, BRM (encoded by SMARCA2) or BRG1 (SMARCC2), three main subunits: BAF155 (SMARCC1), BAF170 (SMARCC2), BAF47 (SMARCB1) and between 7 and 15 additional accessory subunits. Two variants of the complex have been identified and are distinguished based on their subunit composition. BAF (SWI/SNF-A) complexes contain either BA250a (ARID1A) or BAF250b (ARID1B) subunits (figure 2) and Polybromo associated BAF (PBAF) (SWI/SNF-B) complex contain BAF180 (PBRM1) and BAF200 (ARID2) subunits (figure 2).4 ,6 ,11–13

Genomic organisation of SMARCB1 gene. The boxes indicate the coding exons of the gene (numbered 1–9) with the corresponding number of amino acids per exon (above) and gene length, spanning 50 kb (below).

SWI/sucrose non-fermenting (SNF) is involved in chromatin modulation and tumour suppression. Two variants of the complex, BAF and PBAF are shown. BAF and PBAF differ in subunit composition, which are shown in yellow. Core subunits are shown in red, including the ATPase subunits (highlighted with Ben-Day dots). Additional subunits that are encoded by multigene families, which yield in different amino acid sequence, are shown in blue. These complexes make specific contributions to chromatin remodelling and transcription.
Function and mechanisms
SMARCB1 in the SWI/SNF complex plays a critical role in epigenetic regulation, cell cycle progression and crosstalk between signalling cascades.3–6 ,11 ,12 ,14 ,15 First, SMACB1 and the SWI/SNF complex are actively involved in chromatin remodelling and are thought to have both transcription activation and repression roles. ATP-dependent remodelling complexes mobilise the histone-DNA bond using energy generated from ATP hydrolysis and the method of remodelling involve nucleosome sliding, dissociation or replacement (figure 2), although the precise biochemical mechanism remains unknown.4 ,11 ,12 It is also worth noting that the function of SMARCBI within the SWI/SNF complex still remains somewhat nebulous. However, since identification of the SMARCB1 mutation in malignant rhabdoid tumours (MRTs) by Versteege et al 10 there has been some illumination into the role and mechanism of action of the gene. SMARCBI is now recognised as a bona fide tumour suppressor gene, which exerts its tumour suppressor activity epigenetically via transcriptional regulation.3 ,5 ,6 ,12 In addition, SMARCB1 has also been shown to play a role in the downregulating the expression of cyclin-dependent kinase inhibitor p16 (INK4A), Wnt signalling pathway, repressing retinoblastoma (RB) target genes, including E2F factors and CCND1, promote c-Avian Myelocytomatosis Viral Oncogene Homolog Haemoglobin (MYC) oncogene-mediated transactivation, induce aberrant activation of hedgehog (Hh) signalling by interacting GLI1 and localising at GLI1-regulated promoters, and also epigenetically militates the action of the polycomb complex.3 ,5 ,6 ,11 ,12 Some of these pathways will be discussed below and revisited later in this review in the context of targeted therapies.
SMARCB1 in regulation of cyclin D1/CDK4 activation
Studies carried out using MRT cell lines have demonstrated that SMARCB1 represses Cyclin D1 transcription and inhibits the action of cyclin-dependent kinase (CDK4/6) by directly binding and recruiting histone deacetylase (HDAC) activity in G1 of the cell cycle.3 ,11 This then results in hypophosphorylation of RB and induction of P16(INK4A), another tumour suppressor gene3 ,11 (figure 3).

SMARCB1 represses Cyclin D1 transcription and inhibits the action of cyclin-dependent kinase (CDK4) by directly binding retinoblastoma (RB) and recruiting histone deacetylase (HDAC) activity in G1 of the cell cycle, which in turn prevents progression into S phase. RB remains hypophosphorylated, induces release of P16(INK4A) and continues repression of cyclins A and E.
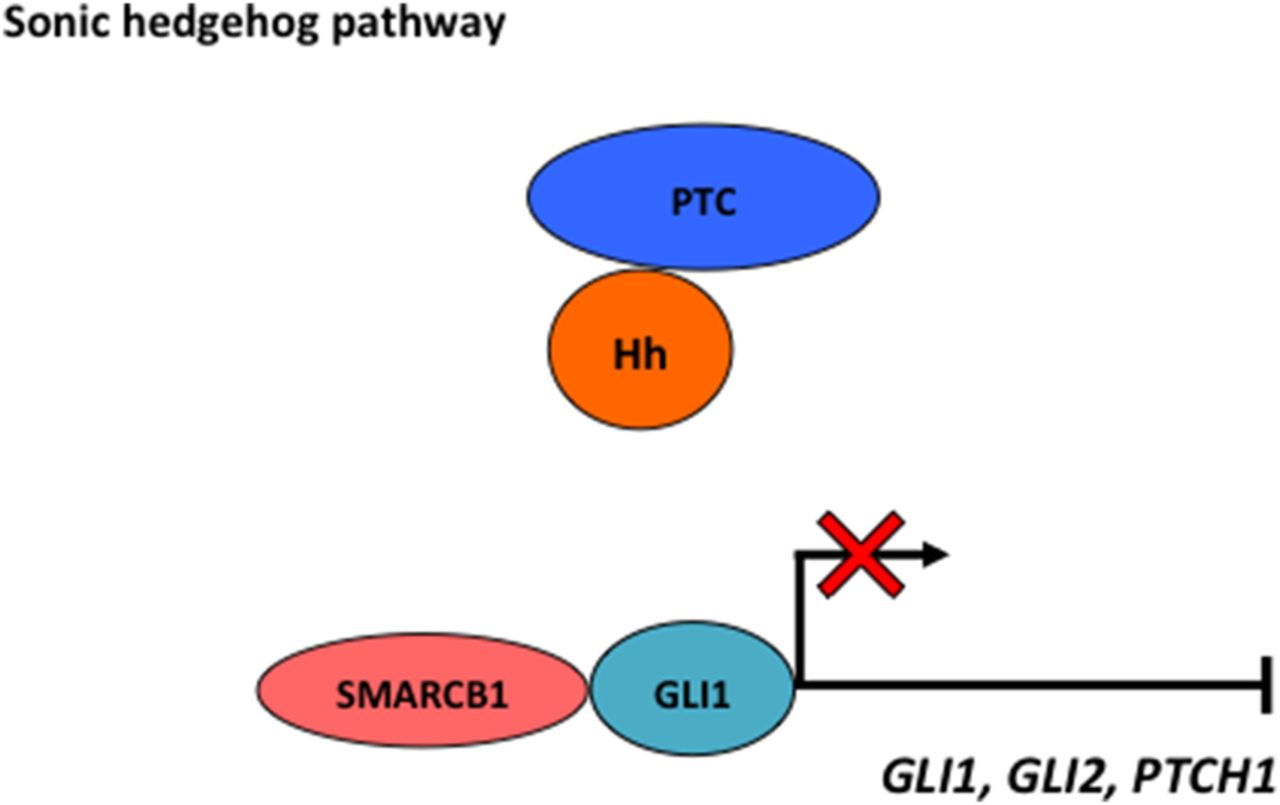
SMARCB1 inhibition of the sonic Hh pathway
The Hh pathway plays important roles in modulating patterning and differentiation during development. SMARCB1 directly prevents transcription of glioma-associated oncogene homologue (GLI), thus resulting in reduced downstream Hh pathway target genes, including GL1, GL2 and protein patched homologue 1 (PTCH1)3 (figure 4).

SMARCB1 directly prevents transcription of glioma-associated oncogene homologue (GLI), thus resulting in reduced downstream hedgehog (Hh) pathway target genes, including GL1, GL2 and protein patched homologue 1 (PTCH1). PTC, patched proteins.
SMARCB1 in regulation of WNT/B catenin pathway, E2F, aurora A and c-MYC
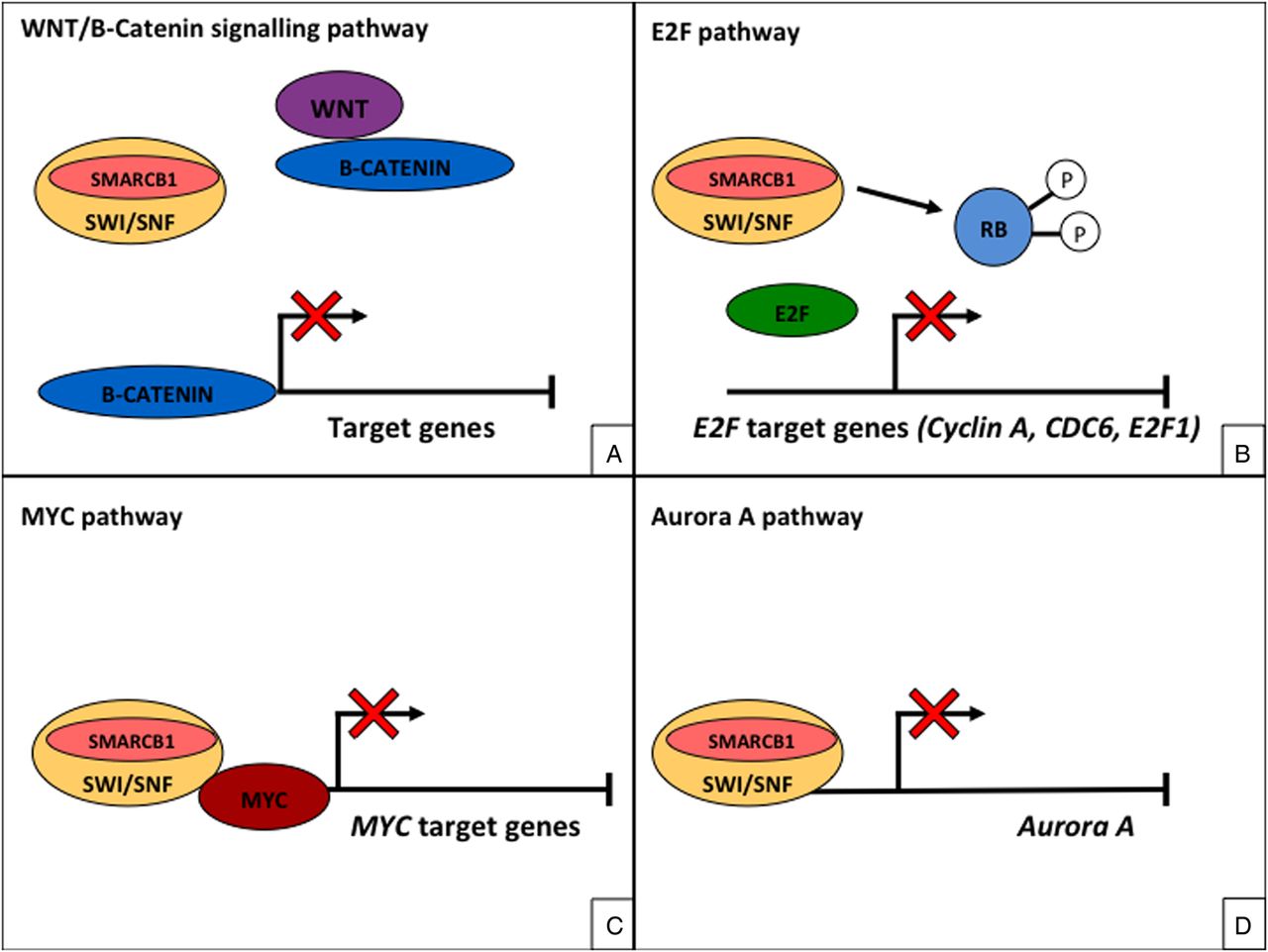
SMARCB1 and the other subunits of the SWI/SNF complex have been implicated in transcriptional regulation of the cell cycle, development, proliferation and differentiation.3 Studies have shown SMARCB1 to play a critical role in regulating the Wnt/B catenin signalling pathway3 ,16 (figure 5A) and several cancer-related genes including E2F-related target genes3 ,17 ,18 (figure 5B). In addition, SMARCB1 may have a role in the expression c-MYC and aurora A3 (figure 5C, D).
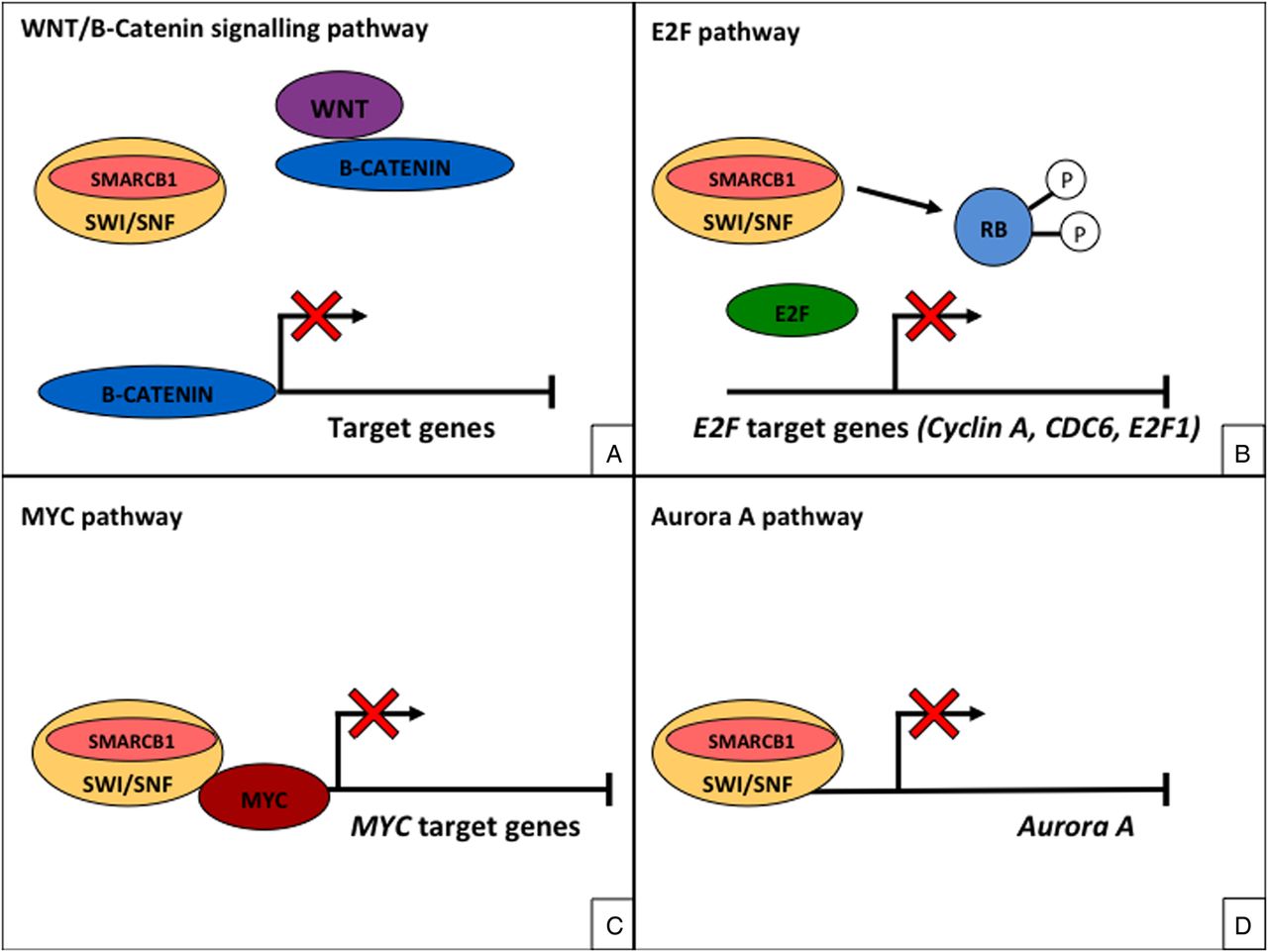
SMARCB1 and the other subunits of the SWI/sucrose non-fermenting (SNF) complex play a critical role in regulating the WNT/B catenin signalling pathway (A) and several cancer-related genes including retinoblastoma (RB)-mediated repression of E2F target genes (B). SMARCB1 may also play a role in the expression c-MYC (C) and aurora A (D).
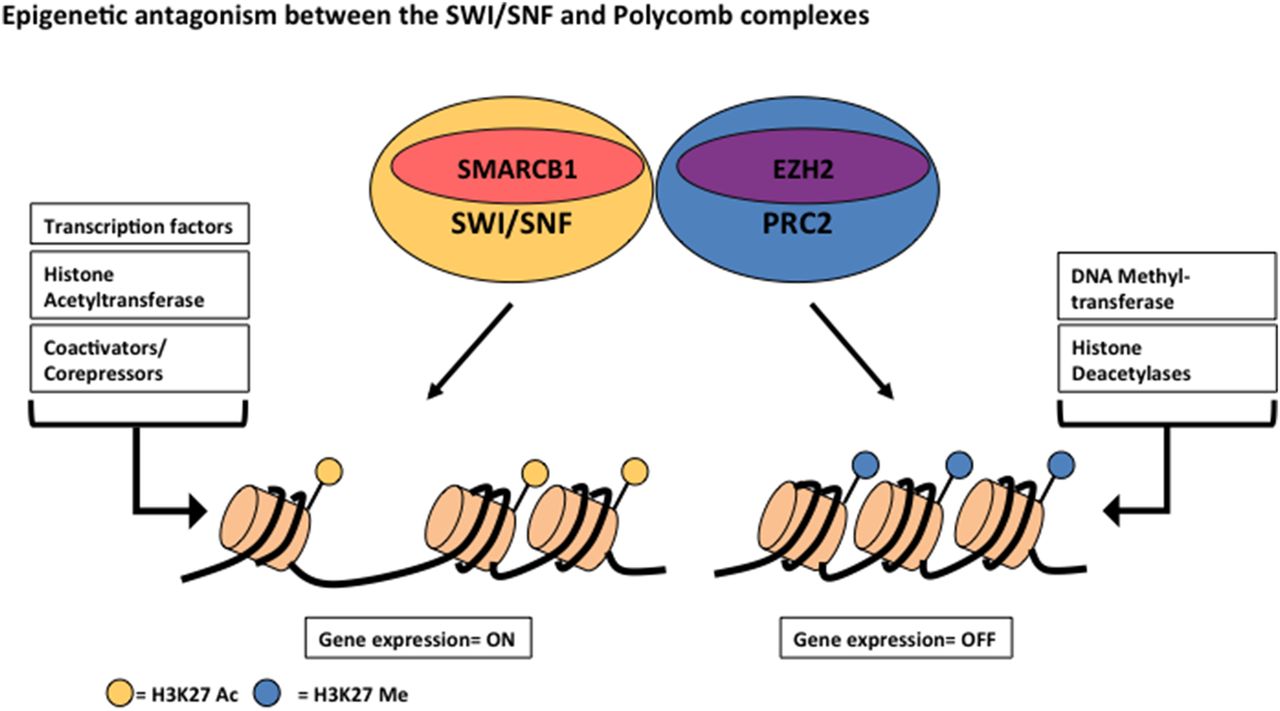
SMARCB1 (SWI/SNF complex) epigenetic antagonism with polycomb complex
Enhancer of zeste 2 (EZH2) is a histone-lysine N-methyltransferase, which is the catalytic subunit of the polycomb repressive complex 2 (PRC2).3 ,5 This complex mediates the trimethylation of lysine 27 of histone H3 (H3K27 Me3), which plays a crucial role in epigenetic silencing and oncogenic transformation.3 As mentioned earlier, PRC2 antagonises the action of SMARCB1, as inactivation of SMARCB1 leads to an elevated expression of EZH2 and histone molecule, H3K27 was found to be present largely in the trimethylated state3 (figure 6).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
During lineage-specific differentiation, the SWI/sucrose non-fermenting (SNF) complex (SMARCB1) interacts with transcription factors, histone acetyltransferases and transcriptional regulators to activate expression of target genes, which results in acetylation of lysine 27 of histone H3 (H3K27 Ac). Antagonising the actions of the SWI/SNF complex is the polycomb repressive complex 2 (PRC2), which contains enhancer of zeste 2 (EZH2). PRC2 interacts with DNA methyltransferases and histone deacetylases to silence gene expression, resulting in methylation of histone H3K27 Me.
SMARCB1 and cancers
Following the identification in MRT,10 SMARCB1 gene abnormalities have also been identified in a variety of other malignant neoplasms (table 1), chiefly epithelioid sarcoma (ES) (conventional and proximal) and renal medullary carcinoma (RMC). In addition, several tumour predispositions syndromes have been linked to SMARCB1 germline mutations (table 1).5 ,12 ,19
SMARCB1 mutations in key tumours
SMARCB1 and MTR
MRTs are aggressive tumours of infancy, which can occur in a wide range of anatomical sites including the kidney, brain (atypical teratoid/rhabdoid tumour), liver, peripheral nerves or soft tissue. These tumours are all characterised by the presence of ‘rhabdoid cells’ featuring large vesicular nuclei, prominent eosinophilic nucleoli and large paranuclear filamentous cytoplasmic inclusion.1 ,12 ,20 Another consistent unifying feature in these tumours is biallelic inactivation of SMARCB1, which is the only gene found to be mutated in ∼95% of all MRTs with unanimous loss of normal nuclear expression of the protein by immunohistochemistry.1 ,3 ,5 ,12 ,14 ,21 The biallelic inactivation of SMARCB1 results in a complete loss of function via a spectrum of events including, whole-gene deletions, large intragenic deletions/duplications, small out-of-frame intragenic deletion/insertions, splice-site mutations and nonsense mutations. Missense mutations are rare. A particular mutation hot spot has not been found; however, exons 1 and 8 are almost never altered.12 Using mouse models, MRTs were established to have a rapid onset and complete penetrance of cancer following inactivation of the SMARCB1,3 with exceptionally low genomic alterations.3 ,12 The absence of genomic instability notwithstanding the rapid onset of the cancer, suggests that the initiation and progression of the cancer may be secondary to epigenetic perturbation of transcriptional regulation.3 ,6
SMARCB1 and ES
Conventional and proximal ESs are distinctive soft tissue tumours of adolescents and young adults. Conventional ES occurs in the distal extremities, the proximal counterpart most often arises in the pelvic and inguinal area and pursues a more aggressive clinical course.2 ,22 Histologically, conventional ES are characterised by nodules of plump epithelioid to spindle cells with central necrosis while the proximal type comprises larger cells with marked atypia, frequently demonstrating a rhabdoid features. Regardless of the type, the majority of ES (80%–90%) show loss of SMARCB1. The absence of protein expression is related to homozygous deletions in most cases,12 ,22 ,23 involving short segments of the gene, either exons 4–5 or exons 6–9.22
SMARCB1 and RMC
RMCs are highly aggressive carcinomas, occurring in young patients carrying sickle cell trait or haemoglobin SC disease.1 ,24 These tumours are thought to arise from renal medullary calyceal epithelium.1 ,24 All cases that have been studied thus far show loss of protein expression of SMARCB1, with loss of heterozygosity at the SMARCB1 locus.1 ,12 ,24 Calderaro et al 24 have suggested that the complete loss of SMARCB1 expression in RMC, in the absence of biallelic inactivation or chromosomal imbalances could be explained by epigenetic silencing acting on the remaining allele.
SMARCB1 and other cancers
The family of tumours harbouring loss of SMARCB1 expression is steadily expanding. Complete loss of SMARCB1 expression has been observed in approximately half of epithelioid malignant peripheral nerve sheath tumours.1 ,20 ,24 Similarly, SMARCB1 expression is lost in up to 40% of paediatric soft tissue myoepithelial carcinomas (MC) and 10% of adult soft tissue MS,1 which may also harbour EWSR1 translocations in ∼50% of cases. However, the underlying genetic alteration in SMARCB1 has not been specified in both the aforementioned tumours.1 ,12 ,20 Occasionally, in the absence of EWSR1/NR4A3 fusion transcripts, truncating mutations, homozygous deletion or microdeletions in SMARCB1 have been reported in extraskeletal myxoid chondrosarcomas.1 ,12 ,25 Cribriform neuroepithelial tumour of the ventricle, an indolent choroid plexus tumour, which shows morphological similarities to RMC, demonstrates consistent loss of SMARCB1 expression and is occasionally associated with biallelic nonsense mutations in SMARCB1. 12 Recently, loss SMARCB1 expression, including homozygous deletions have also been described in undifferentiated chordomas, the more aggressive phenotype of these notochordal tumours of the axial spine.12 ,20 ,26
It is also worth mentioning, a recently recognised group of undifferentiated gastrointestinal carcinomas (UGCs), that demonstrate a morphological spectrum ranging from pure ‘rhabdoid’ features to a poorly differentiated (solid-pattern) adenocarcinoma appearance. These tumours show loss of SMARCB1 loss and are frequently associated with microsatellite instability and loss mismatch-repair proteins as the background genotype.20 ,27 This underpins SMARCB1 loss as a secondary molecular event (double-hit carcinoma). Subsequently, other genes in the SWI/SNF complex, including SMARCA4 and SMARCA2 have been recently found to play a role in the molecular mechanism of UGCs.27
Agaimy et al recently identified a distinctive variant of SMARCB1-deficient sinonasal carcinomas in adults, characterised by superficial papillary fronds, comprising prominent ‘blue’ basaloid cells and scattered rhabdoid cells, which show complete loss of SMARCB1 expression. Importantly, these tumours lack true squamous differentiation, have no detectable human papilloma virus or Epstein–Barr virus DNA and do not harbour an NUT rearrangement, indicating that tumourigenesis is likely related to the SMARCB1 alteration,28 although genetic data on these tumours is presently not available.
Lastly, some studies have shown synovial sarcomas (SSs) to have a reduced SMARCB1 protein expression, which can be demonstrated immunohistochemically as low intensity staining of the tumour nuclei.29 ,30 Kohashi et al 30 posited that SMARCB1 might be regulated through post-transcriptional mechanism via SS18-SSX. Kadoch and Crabtree further elucidated this in a study outlining the mechanism of cellular transformation of SS. They demonstrated that SS18 is a subunit of BAF (SWI/SNF complex) (see figure 1) and in SS, the SS18-SSX fusion protein binds to the BAF complex and competes with the wild-type SS18 in a reversible manner. Integration of the SS18-SSX fusion protein into the BAF complex ejects BAF47 (SMARBC1) protein from the complex. This results in the degradation of BAF47, leading to reduced overall expression of the protein, despite the stable expression SMARCB1.31
SMARCB1 germline mutations
Approximately, 25%–30% of MRTs show germline alterations, irrespective of the anatomical location of the tumour.12 ,19 ,32 All patients who present with two primary tumours are thought to invariably harbour a germline mutation. Germline mutations and deletions have also been identified in pedigrees with multiple affected siblings and have been grouped under, rhabdoid tumour predisposition syndrome 1.12 The spectrum of germline mutations is similar to somatic mutations and include whole-gene deletions, intragenic deletions and duplications, nonsense mutations frameshift mutations; however, missense mutations are almost never identified.19 In most cases with germline mutations, the germline sequences of the parents are normal. This suggests either gonadal mosaicism in one parent or early postzygotic mosaicism in the affected patient.12 ,32
SMARCB1 germline mutations are also responsible approximately 45% of the familial schwannomatosis cases.12 ,14 ,19 Interestingly, the mutations in familial schwannomatosis are predominantly non-truncating splice-site mutations and missense mutations in exon 1, which are usually preserved in MRT.12 ,32 ,33 In addition, the schwannomas usually show a mosaic pattern of SMARCB1 (BAF47) expression in contrast to the complete loss of the protein observed in MRTs. Schwannomatosis-associated mutants consistently retain a normal ability to control cycle functions, representing more hypomorphic mutational variants.12 As such, this could explain the difference in behaviour between these two tumours.
SMARCB1 in genetic disorders
Just to briefly mention, mutations in several genes including SMARCB1 of the SWI/SNF complex have been identified in Coffin–Siris syndrome.19 ,34 This is a rare genetic disorder characterised by developmental delay, severe speech impediment, coarse facial features, hypertrichosis, hypoplastic or absent fifth fingernails/toenails and agenesis of corpus callosum. The mutations commonly identified are either missense or in-frame non-truncating deletions, which is comparable to germline mutations of SMARCB1 in schwannomatosis. Interestingly, affected individuals almost never develop any associated tumours.34
Future targeted therapy
The increasing knowledge on the role of SMARCB1 in epigenetic regulation, cell cycle progression and signalling pathways has provided an ideal platform to trial biologically targeted therapies, particularly in MRT, which still persists to have an overall poor prognosis. The agents currently being trialled are as follows5: (1) EZH2 inhibitors, which prevent MRT progression and have been shown to sensitise tumour cells to the effects of radiation therapy; (2) histone deacetylase inhibitors (HDACi), share a similar mechanism of action to the EZH2 inhibitors; (3) CDK4 inhibitors, inhibit tumour cell growth by G1 arrest; (4) aurora-A-kinase inhibitors, inhibits aurora A and (5) DNA methyltransferase inhibitors.
However, further understanding of the role SMARCB1 gene and its specific function in other tumours will aid in the discovery of new therapeutic avenues in the future.
Take home messages
SMARCB1 is a core subunit of the SW1/sucrose non-fermenting (SNF) ATP dependent chromatin remodelling complex.
SMARCB1 in the SWI/SNF complex plays a critical role in epigenetic regulation, cell cycle progression and crosstalk between signalling cascades.
Involvement of SMARCB1 (in italics) in cancer was first discovered in malignant rhabdoid tumours (MRT) 15 years ago and subsequently seen in a variety of different cancers (ie epitheliod sarcoma, renal medullary carcinoma etc).
Germline mutations in SMARCB1 (in italics) are seen in 24–30% of MRT and are responsible for 45% of the familial schwannomatosis cases.
Further understanding of the SMARCB1 (in italics) gene will aid in the discovery of new therapeutic avenues in the future.
References
Footnotes
Handling editor Cheok Soon Lee.
Contributors Both authors contributed equally to the conception, accrual of information/data and writing of this manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.