Article Text

Abstract
Gastrointestinal stromal tumours (GISTs) are a commonly encountered tumour in routine practice. In the main, the morphology of spindle, epithelioid or mixed are well recognised along with mutations of c-kit. However, there are other genes that are mutated resulting in characteristic clinicopathological correlations. GISTs harbouring platelet-derived growth factor receptor α (PDGFRα) gene mutations lead to a typical morphological constellation of findings: gastric and omental location, gross tumour that is cystic and haemorrhagic, composed of epithelioid, plasmacytoid cells exhibiting pleomorphism, low mitotic count and containing characteristic giant cells with peripherally placed nuclei. These cells are set in a myxoid stroma containing several mast cells. In addition, perivascular/intratumoural hyalinisation is often seen. These tumours are CD117 and DOG-1 positive. GISTs with SDH mutations are multinodular/bilobed/dumb-bell shape tumour masses with mucosal ulceration and histologically characterised by fibrous bands around and within nodules of epithelioid or mixed epithelioid/spindle cells. Lymphovascular invasion with lymph node metastases are usual. Immunohistochemically, the GISTs are CD117, DOG-1 positive, SDHA negative (if SDHA mutated), SDHA positive (if SDHA intact) and SDHB negative. BRAF and NF-1 mutated GISTs do not have any characteristic morphological features.
- GI NEOPLASMS
- MOLECULAR PATHOLOGY
- MORPHOLOGY
Statistics from Altmetric.com
Introduction
Gastrointestinal stromal tumours (GISTs) are now a well-recognised tumour occurring in the gastrointestinal tract and in several extraintestinal locations such as the omentum, mesentery, retroperitoneum, abdominopelvic cavity, soft tissue, uterus, liver, gallbladder, pancreas and the peritoneal serosa. In these locations, GISTs are thought to arise from dispersed interstitial Cajal-like cells that have the potential for bidirectional differentiation towards myoid and neural lines. It is important to ensure that before making a diagnosis of extraintestinal GIST that a connexion to some part of the gastrointestinal tract or a metastatic lesion is first excluded.
The molecular characterisation of GISTs is important as it identifies sensitivity/resistance to receptor tyrosine kinase (TK) inhibitors and hence an important facet of the work-up of these patients. The first assessment of GISTs occurs at a morphological level, and therefore, it behoves the practising pathologist to be aware of the morphological–molecular correlation that impacts on diagnosis and ultimately therapy and prognosis. This mini-review is to focus the attention of the practising pathologist on the clinical and pathological characteristics of a GIST that may help predict the molecular aberration and thus help direct pertinent molecular testing and impact on management strategies.
Morphological and molecular considerations
It is now well known and accepted that there are two basic cell types encountered in GISTs either alone or in combination: spindle, epithelioid or mixed. Furthermore, these two basic cell types may occur in an array of patterns that mimic other mesenchymal neoplasms such as palisaded and interlacing fascicles. There are distinct phenotypic–immunophenotypic and genotypic correlations. Knowledge of these characteristic correlations helps in directing appropriate molecular testing.
There are clearly identified genes that are abrogated in GISTs that allow for a molecular classification (see box 1). Some of the mutations in these genes lead to very characteristic morphological alterations.
Molecular classification of gastrointestinal stromal tumour (GIST)
I. KIT and
II. Platelet-derived growth factor receptor α (PDGFRα) mutated type (80%–90% of GISTs)
KIT/PDGFRα (double) wild-type (10%–15%)
I. Succinate dehydrogenase (SDH)-deficient or negative GISTs/type II (20%–40%)
II. RAS-P (RAS/BRAF) mutant type (15%)
KIT/PDGFRα/SDH (triple) wild-type/type I (1%–2%)
I. NF-1 mutated
II. Sporadic
KIT/PDGFRA/SDH/RAS-P (quadruple) wild-type (5%)
c-kit mutated GISTs
These are the most frequently encountered GISTs. In the stomach they are usually solitary masses with a wide size range, the majority have a spindle cell morphology while some are epithelioid (20%–25%) and a minority is mixed. Those that occur in the stomach tend to have characteristic perinuclear (sometimes intranuclear) vacuoles and often times a palisaded appearance reminiscent of Verocay bodies of a schwannoma. Vascular dilatation and hyalinisation are also present further recapitulating the appearance of a schwannoma. Small intestinal GISTs are usually more frequently composed of spindle cells and have so-called skeinoid fibres which are extracellular (EC) collagen globules.
Immunohistochemistry: these GISTs are CD117, DOG-1 and succinate dehydrogenase (SDH) subtype-B positive.
Other than the fact that c-kit mutations are the most frequent molecular change in GISTs, a spindle cell GIST should invoke the morphological trigger to seek c-kit mutations.
Platelet-derived growth factor receptor α mutated GISTs
Mutations in the platelet-derived growth factor receptor α (PDGFRα) gene account for only 5% of the collective total of c-kit and PDGFRα mutations. Despite its relative rarity, the morphological features of this subset of GIST are extremely characteristic and will guide the pathologist towards recognising this variant especially in unusual locations such as within the abdominal cavity or retroperitoneum.
The gross appearances of PDGFRα mutated GISTs are also very characteristic. They tend to be found in the stomach or omentum, are larger, sometimes multinodular masses with areas of haemorrhage, cystic degeneration and solid areas giving rise to a variegated appearance.
The histological features are highlighted in box 2. The most striking histological feature is that these GISTs are typically composed of epithelioid cells (figure 1). Invariably, they are exclusively epithelioid cells, or rarely may have a minor spindle cell component. The epithelioid cells have abundant eosinophilic cytoplasm round nuclei with vesicular chromatin and a small discernible nucleus. Another typical feature is the plasmacytoid nature of the epithelioid cells with eccentrically disposed nuclei (figure 2). Interspersed among the epithelioid cells are multinucleated cells that range from binucleate to having as many as 7–10 nuclei. The presence of multinucleated cells imparts a pleomorphic low power impression of the GIST. Often times these nuclei are aligned at the periphery of the cell thus retaining the polarity of the nuclei to one end of the cell (figure 3). Rarely, are the nuclei haphazardly arranged and the giant cells that are tocsins of PDGFRα mutated GISTs do not resemble foreign body giant cells that may be encountered around foci of necrosis or degeneration in any GIST. The grossly observed cysts are seen microscopically as cystic structures of varying size and shape (figure 4). The cystic degeneration is an extension of stromal oedema or myxoid change that is seen diffusely throughout the entire GIST. There are accompanying mast cells and occasional lymphocytes in the stroma. Areas of haemorrhage are seen throughout and vary from being frank to more discrete areas with associated haemosiderin and even some of the tumour cells showing evidence of imbibition of haemosiderin. Completing the constellation of very useful morphological features that point towards PDGFRα mutated GISTs are the vessels. Thin-walled vessels that punctuate the lesion are often dilated and their walls are impregnated by fibrin; sometimes organising fibrin thrombi are also noted (figure 5A, B). Lastly, despite the apparent pleomorphism of epithelioid cells juxtaposed with giant cells, PDGFRα mutated GISTs have a low mitotic count and true tumour necrosis (which must be distinguished from haemorrhage and degenerative change) is also not apparent.
Morphological features of platelet-derived growth factor receptor α mutated gastrointestinal stromal tumours
Multinodularity+/−
Epithelioid cells
Plasmacytoid cells
Pleomorphism with giant cells
Myxoid stroma
Mast cells
Perivascular/intratumoural hyalinisation
Cysts and haemorrhage
Low mitotic count

A platelet-derived growth factor receptor α gene mutated gastrointestinal stromal tumour composed exclusively of epithelioid cells.
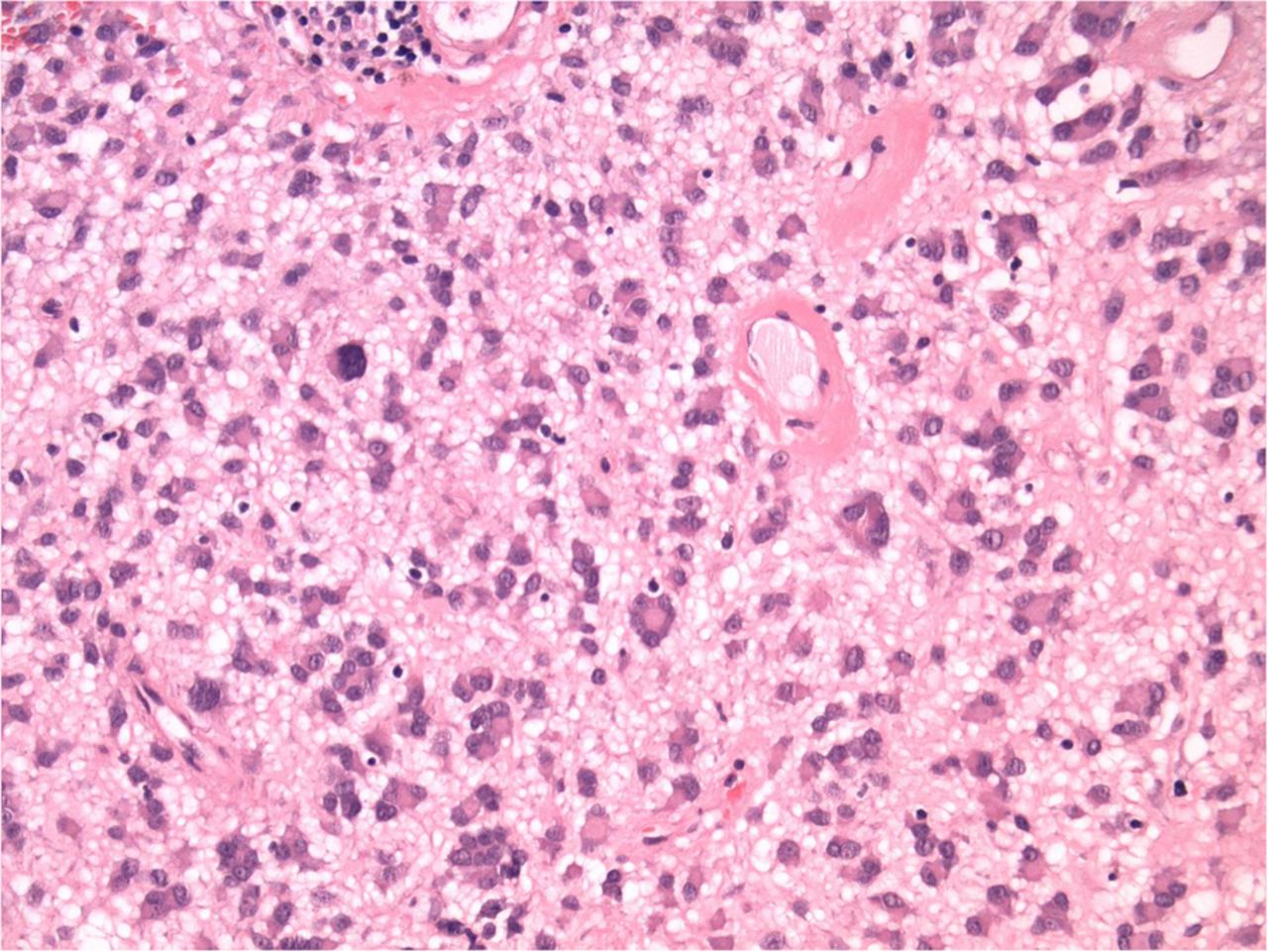
Mild pleomorphism of the epithelioid cells set within a myxoid stroma.

The epithelioid cells have a plasmacytoid appearance and are admixed with characteristic giant cells which range from binucleate to multinucleate.

These gastrointestinal stromal tumours are haemorrhagic and multicystic.

(A and B) Vessels impregnated by fibrin within the walls and showing evidence of organising thrombi.
Mutations in KIT and PDGFRA
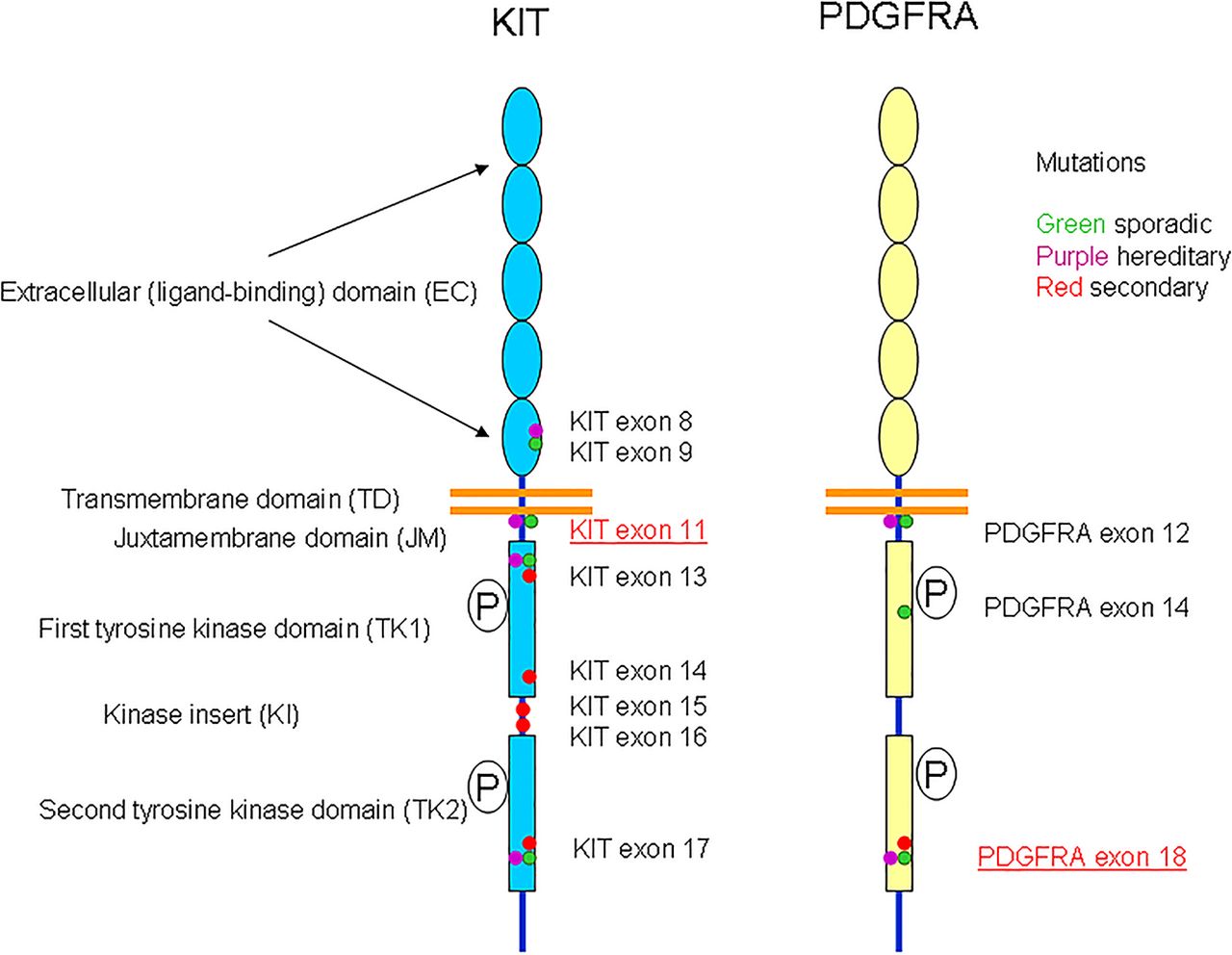
KIT and PDGFRα genes are localised on chromosome 4q12 and might have evolved from a common ancestral gene by gene duplication (see figure 6). They encode a transmembrane glycoprotein that belongs to the type III receptor TK family. KIT and PDGFRA receptors comprise an EC domain with five Ig-like loops and a cytoplasmic domain with juxtamembrane (JM) region and a split TK domain. The TK domain is divided into an ATP binding region (TK1) and a phosphotransferase region (TK2) by a hydrophilic kinase insert (KI) (figure 1). Interaction of the fragment crystallizable (FC) domain with their ligands results in the dimerisation of receptors and phosphorylation of tyrosines in their cytoplasmic TK domains, which leads to a phosphorylation cascade and activation of several signal transduction pathways.1

Schematic representation of the various mutations encountered in KIT and platelet-derived growth factor receptor α (PDGFRα) genes in gastrointestinal stromal tumours.
Mutations in KIT and PDGFRα receptors result in receptor activation secondary to constitutive TKs phosphorylation. Mutations can occur in the receptor regulatory domains (EC and JM) and in the enzymatic domains (TK1 and TK2).
Mutations can be grouped in two categories: primary mutations identified in GISTs before treatment with TK inhibitors, which are linked to tumour pathogenesis and secondary mutations, which are detected during treatment and cause resistance to imatinib-based TK therapy.1
In sporadic GISTs, primary KIT mutation can occur in the EC domain (exon 9), JM (exon 11), TK1 (exon 13) and TK2 (exon17) domains.
Secondary mutation can be found exclusively in the KIT-TK1 (exon 13 and 14) and TK2 (exon 17) and KIT (exon 15 and 16) domains.1
Additionally, imatinib resistance is classified into primary resistance or early progression and secondary resistance or late progression. Primary imatinib resistance is characterised by the lack of initial response with disease progression within 3–6 months of initiating imatinib therapy. In contrast, secondary resistance is characterised by initial response to imatinib and development of resistance within 12–36 months.2
Primary imatinib resistance is found in 50% of PDGFRα-mutant tumours, 33.4% of wild-type tumours and 8.9% of KIT-mutant tumours. KIT exon 9-mutant tumours showed primary resistance more frequently than exon 11-mutant and other tumours.2
Regarding secondary resistance associated with KIT second-site mutations, the exon 17 mutation (54.5%) was most frequent, followed by exon 13 (38.3%) and 14 (13.4%) mutations.2
Total imatinib resistance is seen in 35.5% of PDGFRA-mutant tumours, 33.7% of wild-type tumours (KIT and PDGFRα non-mutant tumours) and 27.4% of KIT-mutant tumours.2
Primary KIT mutations
Deletions: in-frame mutations are the most frequent KIT mutations. They occur almost exclusively in exon 11 and include losses of 3–30 or more nucleotides with deletions or deletion–insertions at the protein level. The most common mutations reported on exon 11 are 1690_1695delTGGAAG (Trp557_Lys558del) and 1755_1759delGAT (Asp579del).
A 2131_2136delAAGAAT (Lys704_Asn705del) in exon 14 in the TK1 domain is the only one deletion found outside the KIT-JM.1
Single nucleotide substitutions are the second most frequent mutations occurring most commonly in exon 11. The most common missense mutations are Val559Asp, Val560Asp, Trp557Arg, Val559Ala, Val559Gly and Leu576Pro.
Rare mutations have also been identified in exon 13; the most common of which, consist of a1945A>G substitution resulting in Lys642Glu at the protein level and in exon 17 which consists of 2487T>A substitutions leading to Asn822Lys at the protein level.1
Duplications have been identified in exon 9 (distal part of KIT-EC domain) and in exon 11 (KIT-JM domain). The majority of exon 9 duplications consists of 1525_1530dupGCCTAT leading to Ala502_Tyr503dup at the protein level.
Duplications in exon 11 are structurally various. Their sizes vary from 1 to 18 codons and cluster in 3′KIT exon 11.1
Insertions are very rare, found only in exon 11 and consists in 1694_1695insTCC leading to Lys558delinsAsnPro at the protein level.
Complex mutations: deletion–insertions and duplication–insertions are relatively rare in KIT exon 11 mutations.
KIT exon 11 deletions, deletion–insertions and single nucleotide substitutions have been identified in GISTs everywhere in the gastrointestinal tract, however, the vast majority (>80%) of KIT exon 11 duplications occur in gastric tumours. KIT exon 11 mutants show more often spindle cell than epithelioid morphology.
The majority of exon 9 duplications have been described in intestinal and rarely in gastric GISTs. Exon 17 mutants occur two times more frequently in small bowel than gastric GISTs, as well as exon 13 mutants GISTs which occur more frequently in the small bowel. KIT exon 9, 13 and 17 mutants often have spindle cell morphology.1
PDGFRα mutations
In sporadic PDGFRα GISTs, primary mutations occur in the JM (exon 12), TK1 (exon 14) and TK2 (exon 18) and secondary mutation in TK2 (exon 18) domain.
Single nucleotide substitutions: most of these mutations have been identified in exon 18 (PDGFRα-TK2). Rarely, single nucleotide substitutions can occur in exon 12 (PDGFRα-JM) and exon 14 (PDGFRα-TK1). In exon 18, the most common is single nucleotide substitution 2664A>T leading to Asp842Val mutation. The second most common mutation in exon 12 is represented by 1821T>A resulting in Val561Asp mutation at the protein level. Mutations in exon 14 are rare and cluster in codon 659.
Deletions: these mutations occur in exon 18 (PDGFRα-TK2) and exon 12 (PDGFRα-JM). They consist of losses of three to several nucleotides and lead to deletion or in some cases deletion–insertions at the protein level.
Duplications are rare and only three such mutations have been identified in PDGFRα exon 12.
Insertions and complex mutations (deletion–insertions): insertions in PDGFRα are extremely rare. Deletion–insertions have been reported in exon 18.
PDGFRα mutations occur almost exclusively in GIST of stomach and omentum.
During imatinib mesylate treatment, resistance often develops due to detected secondary KIT or PDGFRα mutations. Almost all such mutations reported in GISTs affect KIT. Exceptionally, GISTs with primary KIT and secondary PDGFRα, Asp842Val mutations have been reported.
The majority of secondary KIT mutations represent single nucleotide substitutions affecting specific codons in KIT exon 13 and 14 (TK1), exon 15 and 16 (KI) and 17 (TK2). KIT-TK1 domain mutations affect codons never mutated in primary tumours, whereas some of the secondary KIT-TK2 mutations also occur in primary KIT mutations.
SDH-deficient GISTs
GISTs that do not harbour c-kit or PDGFRα mutations are called ‘double wild type GISTs’ and these account for 10%–15% of all GISTs. Accounting for up to 40% of this particular category of GIST are those characterised by SDH deficiency. These GISTs are also called type II or ‘paediatric-like or adult-paediatric’ GISTs. SDH consists of a tetrad of four protein subunits (figure 7): SDH A, B, C and D that play a role in the Krebs' cycle with catalyses the oxidation of succinate to fumarate.

The succinate dehydrogenase (SDH) family consists of a tetrad of subunits.
Briefly, SDH-deficient or negative GISTs display overexpression of insulin growth factor 1 receptor with resultant upregulation of hypoxia-inducible factor 1 and vascular endothelial growth factor causing increased growth signalling. Germline and/or somatic loss-of-function mutations in any of the four constituents of the SDH complex result in this category of GIST. Genome-wide DNA hypermethylation has also been encountered as a cause of SDH deficiency. Of the four subunits, SDHA mutations are the most common (30%) with resultant loss of SDHA and SDHB protein expression immunohistochemically.
Cases with SDHB mutations result in the following immunohistochemical profile: SDHA positive/SDHB negative.
There are clinical characteristics that typify SDH-deficient GISTs. They occur in children, usually a female predilection with an age range 18–30 years, in the stomach (antrum/distal), have an indolent course despite resistance to imatinib. Many of these GISTs arise in two related clinical syndromes. First, the Carney–Stratakis syndrome consists of GIST and paraganglioma and the GISTs are caused by germline mutations in SDHB and SDHC or inactivating mutations in SDHD. Carney's triad (GIST, paraganglioma and pulmonary chondroma), on the other hand, is not associated with SDH mutations.
The morphological features are highlighted in box 3. These GISTs are multinodular or bilobed/dumb-bell shaped masses that may have a mucosal or submucosal component that causes mucosal ulceration. The cell type is usually epithelioid or mixed epithelioid and spindle with bands of fibrous around the nodules and also extending within the nodules of GIST. Lymphovascular invasion and resultant lymph node metastases are common. Immunohistochemically, these GISTs are CD117 and DOG-1 positive SDHA negative (if SDHA mutated) or positive (if SDHA intact) and SDHB negative.
The morphological features of succinate dehydrogenase (SDH)-deficient gastrointestinal stromal tumours
Multinodular/bilobed/dumb-bell shape tumour masses
Fibrous bands around and within nodules
Mucosal ulceration
Epithelioid or mixed epithelioid/spindle
Lymph node metastases
Lymphovascular invasion
KIT, DOG-1 positive
SDHA immunohistochemistry negative, if SDHA mutated
SDHA immunohistochemistry positive, if SDHA intact
SDHB immunohistochemistry negative
Thus, the clinical scenario and the morphology are indicative of SDH-deficient GISTs.
SDHA mutation-associated GISTs
SDH is a heterotetrameric enzyme complex situated in the inner mitochondrial membrane and is entirely encoded by chromosomal DNA. The SDH complex participates in the Krebs' cycle.
In the ‘succinate dehydrogenase deficient’ GISTs, the SDH complex is inactivated in the tumour cells. This may happen via combination of a loss-of-function germline mutations in one of the SDH subunit genes and somatic loss-of-function mutations in the tumour cells, resulting in inactivation of both alleles. In some cases the mechanism of inactivation maybe secondary to an epigenetic event.3
The mutations include frame shift deletions leading to premature stop codons, missense and nonsense mutations and occasionally splice site mutations.3
The most frequently mutated SDH subunit in SDH-deficient GISTs is SDHA. In most cases, these are germline mutations. The most common recurrent SDHA mutation occurs in exon 2 and translates to protein abrogation R31X. Simultaneous allelic loss at the SDHA locus at 5p15 has been detected with comparative genomic hybridisation.3
Mutations of SDHB, SDHC and SDHD are regularly associated with SDH-deficient paragangliomas; however, they seem to occur only in a minority of SDH-deficient GISTs (20%–30%). The majority of these SDH mutations are germline.3
Regardless of the SDH gene status, all SDH-deficient GISTs have a high frequency of gene methylation, in comparison with KIT or PDGFRα mutant GISTs.3
RAS-P mutant GISTs
The BRAF mutation V600E has been found in 3%–13% of c-kit/PDGFRα/SDH or triple wild-type GISTs (figure 8). Overall, BRAF mutations account for 2% of all GISTs. RAS family mutated GISTs include HRAS, NRAS and PIK3CA mutations and these account for <1% of all GISTs. The RAS pathway and BRAF mutated GISTs share similar features although not morphologically distinct at this juncture. They occur in slightly more often in adult females or with equal gender incidence are solitary tumours (not multifocal) and occur most frequently in the small intestine (60%–70%) and then the stomach. These GISTs are CD117, DOG-1 and SDHB positive immunohistochemically.
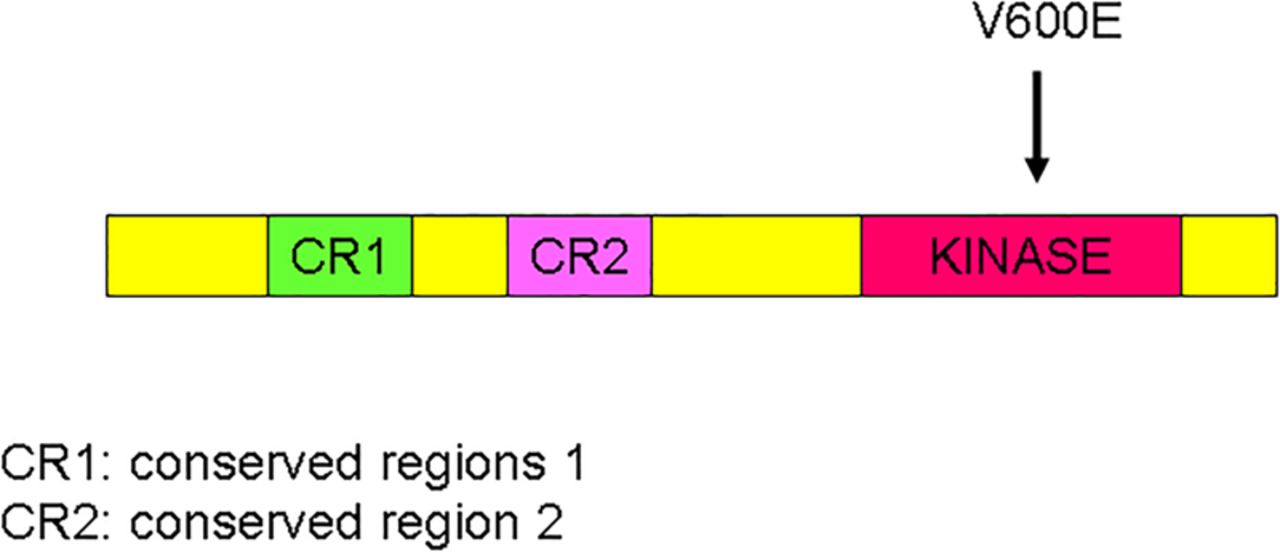
Schematic representation of the BRAF gene with V600E mutation.
BRAF is a member of the Raf kinase family of growth signal transduction protein kinases. This protein plays a role in regulating the mitogen activated protein (MAP) kinase/extracellular-signal-regulated kinases (ERKs) signalling pathway, which affects cell division and differentiation. BRAF is consists of three conserved domains and regulated signal transduction serine/threonine-specific protein kinase.
A BRAF exon 15 mutation (V600E) which results in a constitution activation has been detected in imatinib-naive wild-type high-risk intestinal GISTs (4%–7%). KIT/PDGFRA and BRAF mutations are mutually exclusive. Thus, BRAF mutations represent an alternative molecular pathway in the early tumourigenesis of a subset of KIT/PDGFRA wild-type GISTs. Secondary V600E BRAF mutations may also be an alternative mechanism of imatinib resistance.4 ,5
NF-1 mutated GISTs
Neurofibromatosis 1 (NF-1) mutated GISTs are another example of c-kit/PDGFRα/SDH or triple wild-type GISTs and are related to the RAS pathway mutated GISTs (figure 9). They have an equal gender incidence, occur in young patients and present with multifocal spindle cell GISTs in the small intestine.

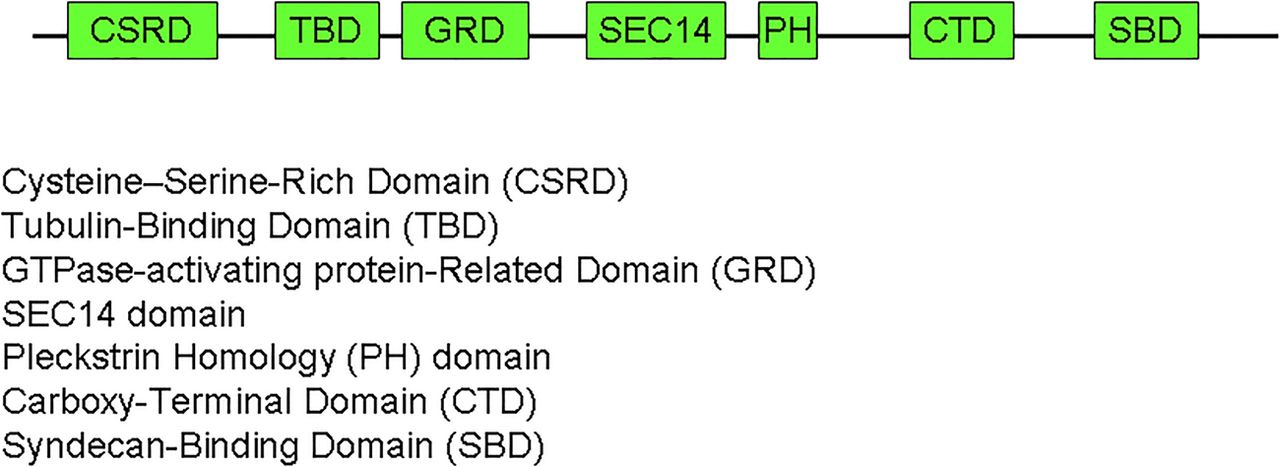
The subunits that constitute the NF-1 gene.
NF-1 is a tumour suppressor gene encoding the protein neurofibromin a member of the RAS regulatory protein family. The NF1 gene is a huge gene of 60 exons located on chromosome 17. It encodes the soluble neurofibromin protein with 2818 residues. NF1 protein consists of a GAP-related domain (GRD) and six potential phosphorylation sites for serine/threonine kinases and one for TKs. NF1 protein acts as a negative regulator of Ras. The GRD of the NF1 protein accelerates the switch of active Ras–guanosine triphosphate (GTP) to an inactive Ras–guanosine diphosphate (GDP). The NF1 protein has also been shown to interact with cytoskeleton and to be involved in the adenylyl cyclase/protein kinases A (PKA) pathway.6
NF-1 mutations result in a production of a short form of neurofibromin. KIT and PDGFRα mutations are very rare events in NF-1 mutated GIST and when present are not believed to be causal. They may be a late event in the tumour development, not an essential event. It is possible that the inactivation of the NF-1 tumour suppressor pathway plays an alternative direct role in the pathogenesis of this subgroup of GISTs. Inactivation of the NF1 gene leads to a constitutive activation of the Ras–mitogen activated protein kinase (MAPK) pathway, which may play an important role in cell proliferation, dysregulated cell growth and tumourigenesis of NF-1 GISTs. NF1 may also be associated with microtubules and modulate the cyclic adenosine monophosphate (CAMP)–PKA signalling pathway.7 Additionally, loss of heterozygosity (LOH) at 14q and 22q has been identified and may contribute to the relatively early phase of tumour development of NF-1 mutated GIST,8 due to loss of tumour suppressor gene on these chromosomes.
Unknown mutated GISTs
These represent c-kit/PDGFRα/SDH/RAS-P (quadruple) wild-type GISTs and overall account for 5% of all GISTs. As these represent the rarest of all the categories, not enough is known about the morphology.
A whole genome analysis showed that the quadruple negative tumours have a genomic expressing signature extremely different from both either KIT/PDGFRα mutated or SDHA mutated GIST. These quadruple negative GISTs are characterised by overexpression of molecular markers (calcitonin receptor like (CALCRL) and collagen type XXII alpha 1 (COL22A1)) and of oncogenes including tyrosine and cyclin-dependent kinases (neurotrophic receptor tyrosine kinase (NTRK2) and cycling dependent kinase (CDK6)) and one member of the erythroblast transformation- specific (ETS)-transcription factor family (ETS-related gene ERG). This would suggest that this is a unique subset of GISTs.9
Conclusions
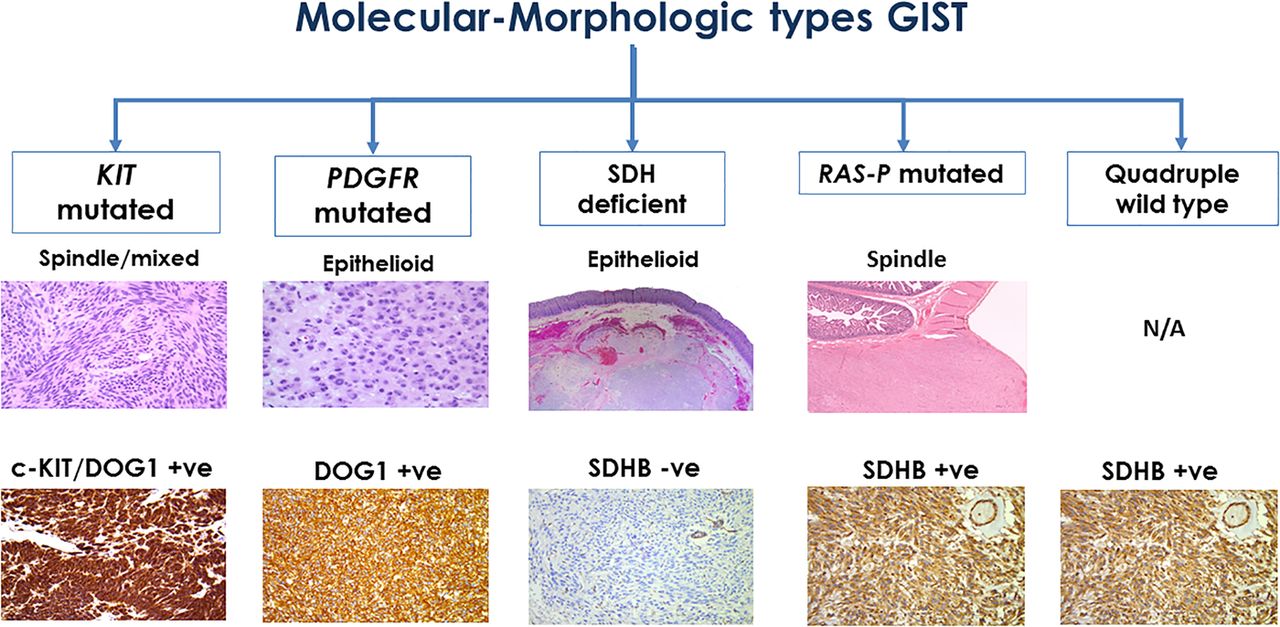
There are well established molecular–morphological correlations in GIST (figure 10). Awareness of morphology helps predict the molecular alteration in several (not all) GISTs. These characteristic associations assist the pathologist in ordering pertinent molecular tests which assist in determining response to therapy.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
An algorithm demonstrating the morphological and molecular correlations in GISTs. GIST, gastrointestinal stromal tumour; PDGFR, platelet-derived growth factor receptor; SDH, succinate dehydrogenase.
Take home messages
The vast majority of GISTs are c-kit or PDGFRα mutated.
Most c-kit mutated GISTs are spindled and/or epithelioid.
PDGFRα mutated GISTs are epithelioid, myxoid and have multinucleated giant cells.
SDHB immunohistochemistry should be performed, especially in multinodular or bilobed GISTs.
Footnotes
RC and SS contributed equally.
Handling editor Cheok Soon Lee
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.