Article Text

Abstract
Accurate diagnostic interpretation of a lymphoid population composed predominantly of small T cells, together with smaller numbers of large B cells, with or without a nodular architecture, is a common problem faced by the histopathologist. The differential diagnosis of this histological pattern is wide, ranging from reactive conditions such as drug reactions and viral infections, through borderline entities such as immunodeficiency-related lymphoproliferative disorders to lymphomas. The latter includes entities where the large B cells are primarily neoplastic (classical and nodular lymphocyte-predominant Hodgkin lymphomas and T cell/histiocyte-rich large B cell lymphoma) as well as T cell lymphomas such as angioimmunoblastic T cell lymphoma where the large B cells represent an epiphenomenon and may or may not be neoplastic. Several rare variants of these conditions, and the fact that treatment can significantly modify appearances, add to the diagnostic difficulty of these pathological entities. Unlike monomorphic lymphoid infiltrates, the histological pattern of T cell-rich proliferation with large B cells requires close evaluation of the inter-relationship between B cells and T cells, follicular dendritic cells and sometimes other inflammatory cells. Epstein-Barr virus plays a key role in several of these scenarios, and interpreting not only its presence but also its distribution within cellular subgroups is essential to accurate diagnosis and the avoidance of some important diagnostic pitfalls. An understanding of normal immunoarchitecture and lymphoid maturational pathways is also fundamental to resolving these cases, as is a knowledge of their common patterns of spread, which facilitates correlation with clinical and radiological findings.
- HAEMATOPATHOLOGY
- HISTOPATHOLOGY
- HODGKINS DISEASE
- LYMPH NODE PATHOLOGY
- LYMPHOMA
Statistics from Altmetric.com
Introduction
Although changing lifestyle and environmental factors have increased the relative proportion of carcinomas among malignancies worldwide in the past decade, lymphoma remains in the top 10 most common malignancies in terms of global incidence.1 Outside the context of staging for metastasis, the most important indication for lymph node biopsy is the investigation of lymphoproliferative disorders (LPDs). Many of these manifest histologically as a monomorphic cellular infiltrate, thus often making the diagnosis straightforward. A number of lymphoproliferations share a common broad histological pattern characterised by a biphasic appearance, with a predominant population of small T lymphocytes admixed with a smaller population of larger B lymphoid cells. We outline here a diagnostic approach to this histological scenario, including potential diagnostic pitfalls, to help guide accurate identification of specific disease entities.
Low-power morphological assessment of the lymph node architecture is the first essential diagnostic step in characterising these biphasic lymphoid populations. The presence or absence of architectural effacement, usually manifested as loss of sinuses and normal lymphoid follicles, should be noted; effacement by a cellular infiltrate causing loss of these structures, as opposed to their displacement by a reactive expansion of follicles or paracortex, strongly suggests lymphoma. Whether there is significant spillover of the lymphoid population beyond the nodal capsule should also be assessed; lymphocytes are mobile cells that may spread beyond the lymph node as part of florid-reactive hyperplasia, but should not do so in large numbers in benign conditions. The presence or absence of large lymphoid nodules also needs to be determined, as well as whether, if present, these represent expanded true follicles or merely cellular nodules lacking a relationship to follicular structures. The use of markers for follicular dendritic cell (FDC) meshworks (eg, CD21 and CD23) helps elucidate whether the nodules relate to pre-existing follicles or not. Accurate characterisation of the lineage of the lymphoid population is crucial and immunohistochemistry for CD3 and CD20 is valuable for the identification of B-cells and T cells, within some limitations. CD3 is a sensitive and specific marker for T-lymphocytes but is also expressed by natural killer cells and, counterintuitively, is negative in up to 15% of cases of peripheral T cell lymphoma (PTCL).2 The addition of CD2 may help where CD3 is lost. CD20 stains the vast majority of B lymphocytes but is negative in the most immature cells (lymphoblasts) and also, at the opposite end of the maturational spectrum, in plasma cells. CD20 expression is also usually absent in classical Hodgkin lymphoma (CHL) and may be suppressed by anti-CD20 therapy. It is therefore advisable to use PAX5 as an adjunctive B cell marker to highlight CD20 negative B cells, the exception being in plasmacytic/plasmablastic infiltrates where both markers are likely to be negative. The addition of CD79a helps cover the latter spectrum.3 The entities that the pathologist should keep in mind as causes of a T cell-rich lymphoid infiltrate with large B cells are presented in table 1.
Entities that should be considered as causes of a T cell-rich lymphoid infiltrate with large B cells
Discussion of key entities
Reactive: infectious mononucleosis
Infectious mononucleosis (IMS) is a clinical syndrome most commonly due to primary Epstein-Barr virus (EBV) or, more rarely, cytomegalovirus infection, and usually occurring in adolescents and young adults.4 Patients may experience fever, fatigue and sore throat and present with enlarged tonsils, cervical lymphadenopathy, hepatosplenomegaly and circulating atypical lymphocytes. The latter represent CD8-positive cytotoxic T cells produced in response to the infection.4 ,5 Most patients are diagnosed clinically, with laboratory confirmation by positive heterophile antibodies (monospot test) and serological tests like IgM and IgG for EBV viral capsid antigen. Immunoglobulin assays are more reliable and specific than monospot testing.4 ,6 In IMS, EBV primarily infects B-lymphocytes.7
Infectious mononucleosis
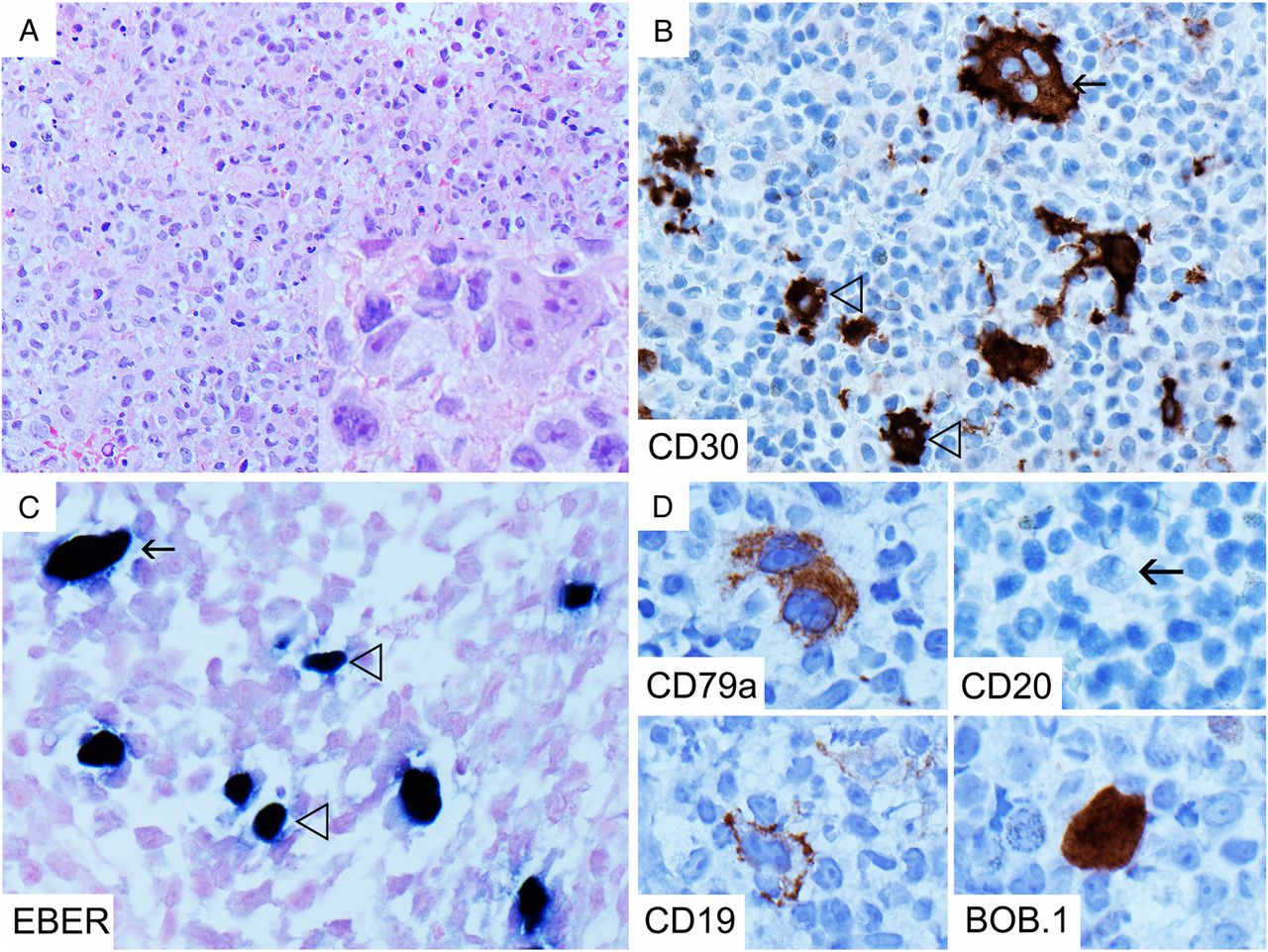
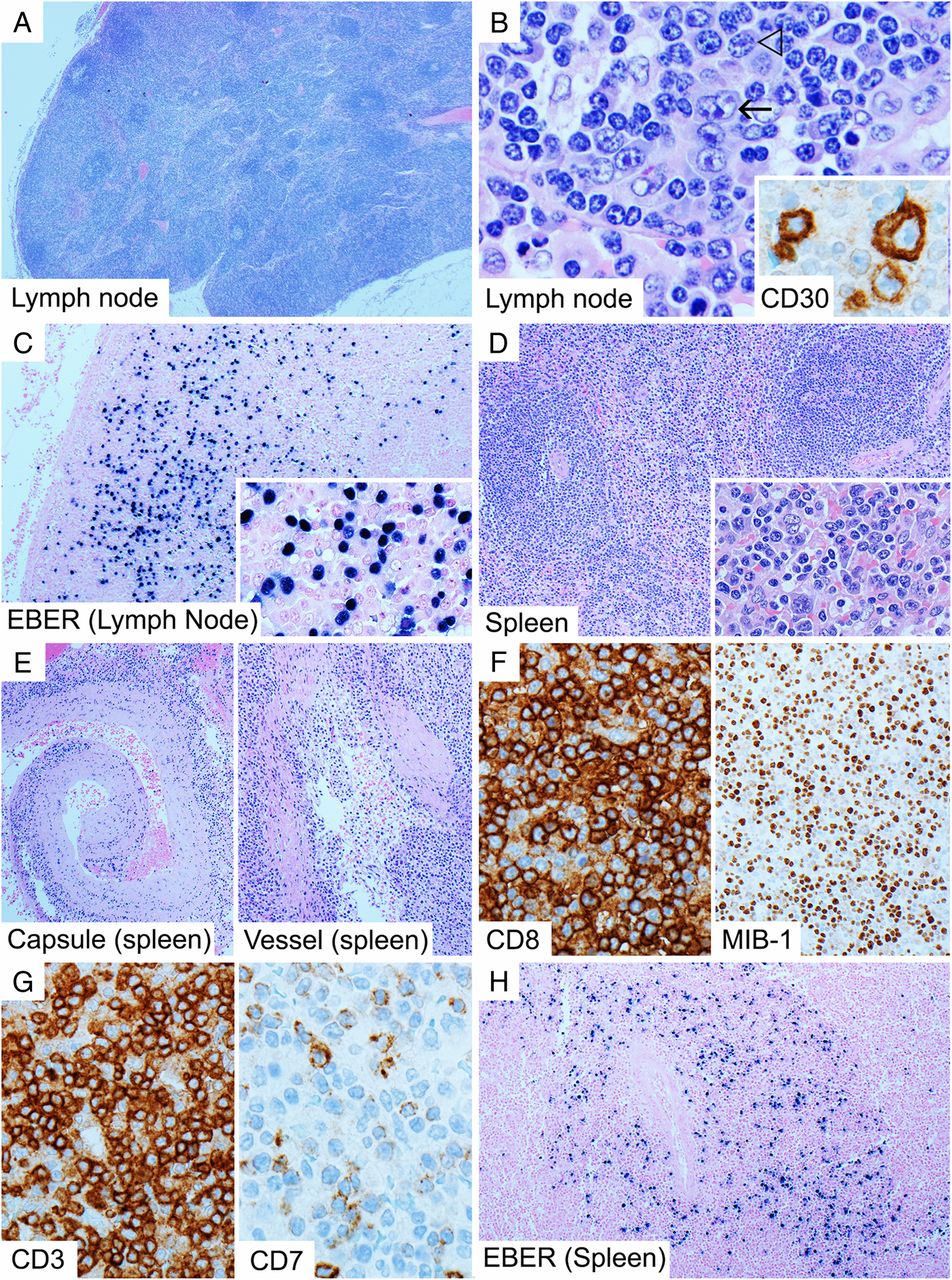
Histopathological misdiagnosis in IMS usually occurs when appropriate clinical details are not received or the condition is not clinically suspected. In lymph nodes and tonsils, there is follicular and paracortical hyperplasia, with a prominent immunoblastic reaction, usually of activated B-immunoblasts, which frequently express CD30 in varying degrees (figure 1B, inset), intermingled with plasma cells and predominantly cytotoxic T cells.5 ,6 ,8 ,9 Immunoblasts may occur in sheets and show high proliferative activity and necrosis, sometimes resulting in a resemblance to diffuse large B cell lymphoma (DLBCL).5 ,9 Another potential diagnostic pitfall is that the CD30-positive immunoblasts can sometimes resemble Hodgkin/Reed-Sternberg (HRS-like) cells (figure 1B), and coupled with a loss of CD45/leucocyte common antigen (LCA) and B cell markers and transcription factor expression that sometimes occurs in these cells,5 ,8 ,10 ,11 this can result in misdiagnosis of CHL.5 ,6 ,9

Lymph node from a patient with infectious mononucleosis showing paracortical expansion but general preservation of architecture (A, H&E 20×) as well as Hodgkin/Reed-Sternberg-like cells (B, H&E 600×, arrow), although more typical appearing immunoblasts are also present (B, H&E 600×, open arrow head). The immunoblasts are CD30 positive and show some variation in size (B, inset, CD30 600×). There are numerous Epstein-Barr virus by in situ hybridisation for EBER (EBER-ISH)-positive cells, including small-sized and medium-sized cells (C with inset, 100× and 400×). The spleen from the same patient shows expanded white pulp follicles without germinal centres and red pulp infiltrated by atypical activated lymphocytes (D with inset, H&E 100× and 400×), which infiltrate the ruptured splenic capsule and vessels (E, H&E, 100×). Many of these are CD8-positive T cells (F, 400×) which show high proliferative activity (F, MIB-1, 200×) and loss of CD7 compared with CD3 (G, 400×). There are numerous EBER-ISH-positive cells in and around the white pulp follicles (H, 100×).
It is therefore critical to recognise the preservation of a normal overall architecture (figure 1A) and the heterogenous cytological appearance of the immunoblasts that typify IMS.5–7 ,9 HRS-like cells in IMS usually occur in a wide range of cell sizes and accompany more typical appearing immunoblasts (figure 1B), in a spectrum differentiating towards plasma cells, which is not typical of CHL, where the neoplastic cells tend to be more homogenous.6 Furthermore, the HRS-like cells of IMS usually retain some expression of B cell markers unlike true HRS cells.6 ,7 ,9 A particularly useful diagnostic feature is the widespread positivity for EBV by in situ hybridisation for EBER (EBER-ISH) (figure 1C) within B cells, which is otherwise rare outside the context of Burkitt lymphoma and EBV-positive DLBCL. The presence of EBV positivity in both large and small lymphocytes distinguishes it from EBV-positive DLBCL and CHL.5 ,6 Furthermore, CD15 is usually negative in the B-immunoblasts.5 ,6 ,9 ,11 There is however no substitute for close correlation with the clinical presentation and results of serological investigations.6 In children and adolescents, it is especially important to exclude the possibility of IMS prior to diagnosing DLBCL.5
More challenging is the histological appearance of extranodal manifestations of IMS.12 An example is the spleen, which may be received when splenic rupture occurs. Here, white pulp follicles usually lack germinal centres and the red pulp is diffusely infiltrated by medium to large sized, often atypical-appearing, activated lymphocytes (figure 1D) that also infiltrate the vessel walls and capsule (the latter causing the propensity to rupture) (figure 1E).13 ,14 These correspond to the circulating CD8-positive T cells, may be highly proliferative (figure 1F) and often show downregulated expression of CD7 (figure 1G).15 ,16 Due to the intensity of the cytotoxic T cell response, T cell clonality studies may show oligoclonal or clonal T cell receptor gene rearrangements,5–7 ,15 ,17 which may mislead the pathologist to diagnose T cell lymphoma. As in lymph nodes, however, the activated lymphoid population in IMS is polymorphic and includes frequent EBV-infected B-immunoblasts in addition to T cells (figure 1H). The list of diagnostic points and pitfalls is summarised in Table 2.
Borderline: immunodeficiency-related LPDs—polymorphic (including Hodgkin lymphoma-like)
Immunodeficiency states, including primary immunodeficiencies, post-transplant and other iatrogenic conditions (eg, immunosuppressive treatment) and HIV infection, are associated with an increased risk of LPDs.3 Post-transplant lymphoproliferative disorders (PTLDs) are among the best characterised, the majority being of B cell origin and usually associated with EBV infection.18 ,19 Both lymph nodes and extranodal sites may be involved.3 As well as non-Hodgkin's lymphoma and CHL subtypes morphologically similar to those in immunocompetent patients, immunodeficiency-related lymphoproliferative disorders (ID-LPDs) include polymorphic lymphoproliferations that are relatively specific (though not exclusive) to the immunosuppressed state.3 ,18 ,20 ,21 Polymorphic ID-LPD, in particular, may produce the appearance of small T cells together with smaller numbers of large B cells.
Immunodeficiency-related lymphoproliferative disorders (ID-LPDs): polymorphic and Hodgkin lymphoma-like
Polymorphic ID-LPD is most frequently seen as a PTLD, although it can occur in any of the above-mentioned immunodeficiency states.3 ,18 ,21 ,22 There is usually a polymorphic population of immunoblasts, small-to-medium-sized lymphocytes and plasma cells, associated with architectural effacement (figure 2A–C) and/or extranodal infiltration, sometimes accompanied by necrosis and increased proliferative activity.3 ,18 Clinically and radiologically, these lesions are frequently destructive of tissue but yet not histologically typical of lymphoma due to their polymorphic appearance and a defining absence of significant atypia. It is important to note that a bona fide lymphoma may have diffuse atypical cells that appear polymorphic, but with a degree of atypia sufficient to classify them as a lymphoma; these should not be regarded as polymorphic ID-LPD. EBER-ISH-positive cells are usually frequent in polymorphic ID-LPD (figure 2C).3 ,20 A lack of clinical information regarding an underlying immunodeficiency state is often a problem in reaching a diagnosis of polymorphic ID-LPD, except in the post-transplant setting where patients are often under active surveillance for PTLD. Immunohistochemistry in polymorphic lesions should not be interpreted in isolation of the morphology as patterns of staining may sometimes be very similar to lymphoma (figure 2D). It is however important to ensure that sufficient work-up of the T cell population is performed to exclude a T cell lymphoma since the latter are often polymorphic in appearance.20 Even when immunoglobulin light chain restriction is not demonstrable on immunohistochemistry, polymorphic ID-LPD commonly shows clonal immunoglobulin gene rearrangement.3 ,20 The importance of distinguishing PTLD and immunosuppressant-related LPD from other lymphomas is that reduction in immunosuppression treatment often forms part of the first-line treatment.18 ,19

Immunodeficiency-related polymorphic lymphoproliferative disorder associated with methotrexate treatment for rheumatoid arthritis: polymorphic population of immunoblasts with small-to-medium-sized lymphocytes and plasma cells in a lymph node (A, H&E 100× with inset 600×). The lymph node architecture is effaced, with only a few remnant follicles at the periphery highlighted by CD21 immunostaining for follicular dendritic cells (open arrow head) (B, 20×). The infiltrate has a T cell-rich area as shown by CD3 immunohistochemistry (B, inset, 400×), and the B-immunoblasts are negative for CD3 (arrow). The B-immunoblasts are highlighted by CD30, CD20 and some are Epstein-Barr virus by in situ hybridisation for EBER (EBER-ISH) positive (C, 600×). The same lymph node also contains cells within the polymorphic infiltrate showing strong PD-1 positivity, but there are no other features to suggest an angioimmunoblastic or other peripheral T cell lymphoma (D, 100×).
Some cases of polymorphic PTLD can show large immunoblasts that are HRS-like. These are categorised as ‘Hodgkin lymphoma-Like’ (HL-like) PTLD (figure 3A).22–25 HL-like PTLD was previously classified together with post-transplant-related CHL but has since 2008 been regarded as within the spectrum of polymorphic PTLD.3 ,20 ,24 Some authors have observed that HL-like PTLD behaves more aggressively than other polymorphic PTLDs.23 ,25 HL-like PTLD is distinguished from CHL by the greater heterogeneity in the size of the cells positive for EBER-ISH and CD30 (figure 3B, C),23–25 in contrast to CHL where only the large HRS cells are positive for CD30 and EBER-ISH. In addition, while CD45/LCA expression can be variable, CD15 is usually negative and B cell markers/transcription factors like CD79a, CD20, PAX5, Oct-2 and BOB.1 (figure 3D) are more frequently expressed.23–25 However, some patients may have received prior anti-CD20 treatment for previous or suspected PTLD, which can result in CD20-negative HRS-like cells (figure 3D).18 The list of diagnostic points and pitfalls is summarised in (Table 3).
Polymorphic Hodgkin lymphoma-like post-transplant lymphoproliferative disorders post anti-CD20 treatment: This lymph node shows a polymorphic infiltrate with areas of incipient necrosis (A, H&E 100×), as well as Hodgkin/Reed-Sternberg (HRS)-like cells (A, H&E, inset 600×). CD30 immunohistochemistry highlights the HRS-like cells (arrow) and smaller accompanying immunoblasts (open arrow head) (B, 400×). EBER also highlights a mixture of large cells (arrow) and smaller cells (open arrow head), which would be unusual for classical Hodgkin lymphoma (C, 600×). HRS-like cells are also positive for B cell markers and transcription factors like CD79a, CD19, BOB.1 (D, 600×) and Oct-2 (not shown), indicating retention of the B cell programme, but are negative for CD20 (arrow), due to the recent anti-CD20 treatment (D, 600×).
Neoplastic T cells: angioimmunoblastic T cell lymphoma
The occurrence of large B cells within a T cell-rich background is well described as a manifestation of PTCL26 ,27 and angioimmunoblastic T cell lymphoma (AITL) is the entity most closely associated with this finding.28 Occasionally, large B cells occur as confluent sheets amounting to DLBCL.29 AITL is most frequently nodal in origin, but often shows extranodal spread (eg, spleen, bone marrow), and has a characteristic clinicopathological presentation.30 ,31 This often includes generalised lymphadenopathy, constitutional symptoms and hypergammaglobulinaemia, as well as hepatosplenomegaly and skin rashes.32 ,33 Histologically, lymph nodes show variable degrees of architectural effacement by a polymorphic population of lymphocytes, plasma cells and eosinophils associated with an increase in vascularity (figure 4A).31 One of the most characteristic architectural features is expansion and hyperplasia of the FDC meshworks (figure 4B), which encircle high endothelial venules (HEVs).34 There are three histological patterns of AITL based on their follicular architecture, namely pattern 1 (associated with hyperplastic follicles), 2 (regressed follicles) and 3 (effaced follicles).31
Angioimmunoblastic T cell lymphoma
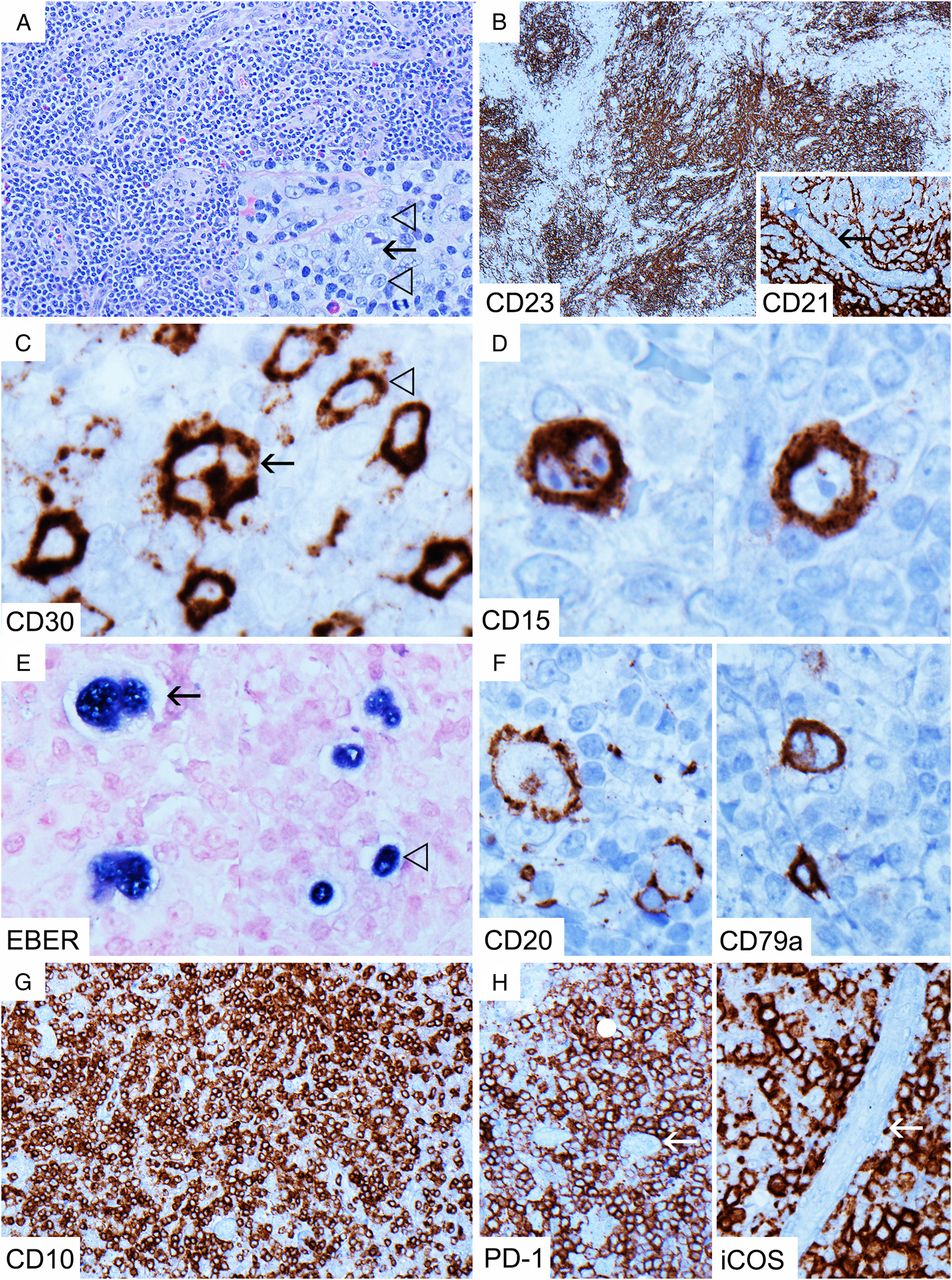
Angioimmunoblastic T cell lymphoma: polymorphic infiltrate with increased vascularity (A, H&E 200×). Large B cells resembling Hodgkin/Reed-Sternberg (HRS)-like cells may be present (A, inset, arrow, H&E 600×). However, note the adjacent medium-sized atypical T cells (A, inset, open arrow head, H&E 600×). There are hyperplastic follicular dendritic cell meshworks (B, CD23 40×) encircling high endothelial venules (HEVs) (arrow) (B, inset, CD21 400×). HRS-like cells are positive for CD30 (arrow) (C, 600×), CD15 (D, 600×) and Epstein-Barr virus by in situ hybridisation for EBER (EBER-ISH) (arrow) (E, 600×), as well as B cell markers CD20 and CD79a (F, 600×). There are also smaller medium-sized cells positive for CD30 (C, open arrow head, 600×) and EBER-ISH (E, open arrow head, 600×), which would not be present in classical Hodgkin lymphoma. Neoplastic T cells are positive for follicular helper T cell (TFH)-associated markers CD10 (G, 200×), PD-1 (H, 200×) and iCOS (H, 400×), and the TFH cells appear to concentrate around HEVs (arrow) (H, PD-1 and iCOS).
Among the diagnostic challenges of AITL is the fact that the neoplastic T cell population may be in a small minority and/or only mildly atypical, leading to the T cell infiltrate being interpreted as reactive,31 ,35 Additionally, the large B cells present in AITL may be HRS-like (figure 4A, inset), leading to a misdiagnosis of CHL.33 ,36 The situation is further complicated by the fact that large B cells in T cell lymphomas are commonly CD30 positive (figure 4C), may express CD15 (figure 4D) and can show variable attenuation of immunostaining for B cell markers/transcription factors, further mimicking HRS cells.33 ,36 ,37 EBER-ISH, though usually positive in the large B cells (figure 4E), is negative in up to 20% of cases.33 ,34 ,36
Fortunately, there are several useful morphological and immunoarchitectural clues to suggest AITL. In typical cases, the background polymorphous infiltrate includes increased numbers of medium-sized cells, representing the neoplastic T cells, some of which may show a clear cytoplasm.34 ,36 Their presence reduces the perceived size contrast between the background small lymphocytes and large B cells, producing a spectrum of cellular sizes not typical of CHL. Large B cells also tend to show expression of more B cell markers than usually present in CHL (figure 4F).36 In AITL patterns 2 and 3, FDC meshworks are often dramatically increased (figure 4B) in an irregular tentacular fashion and encircle HEVs,34 which is not a feature of CHL. CD23 can sometimes be more effective than CD21 in highlighting these meshworks.33 The atypical T cells often show loss of T cell markers, most often CD7 and CD5,2 ,28 ,38 and express CD4, CD10 (figure 4G)31 and follicular helper T cell (TFH) markers including PD-1, iCOS, BCL6 and CXCL13 (figure 4H), especially around HEVs.33 ,39–42 To confirm TFH phenotype, it is advisable to demonstrate positivity for at least two or three TFH associated antigens (including CD10 and BCL6).39 ,43 PD-1 is generally more sensitive than CD10.44 T cell clonality helps support the diagnosis, although this is absent in approximately 10% of cases.34 ,45 ,46 It should be noted that B cell clonality may sometimes be seen in the absence of histological evidence of DLBCL28 ,29 ,46 and by itself is not an indication of concurrent B cell lymphoma.26
As mentioned, AITL can present with hyperplastic follicles (pattern 1), a feature often seen in early/partial lymph node involvement by AITL.29 ,32 ,33 ,45 This pattern is generally associated with a better prognosis,45 although patients with this pattern may still have disseminated disease.29 ,32 ,33 Differentiating AITL pattern 1 and reactive lymphoid hyperplasia can be challenging, and the diagnosis of AITL essentially hinges on the characteristic polymorphic cellular infiltrate described above, and the presence of perifollicular/extrafollicular T-lymphocytes expressing CD10 and TFH markers, because FDC meshwork expansion may be subtle.32 ,38 Features against AITL and in favour of reactive hyperplasia include a uniform expression of CD10 within germinal centres, a low interfollicular proliferation fraction and a young age.38 The pathologist should however exercise caution when interpreting PD-1 and iCOS as these markers, while sensitive, are not entirely specific for TFH.37 ,47–49 PD-1 is normally intensely expressed in the TFH cells within the light zone of the germinal centre, which serves as a reference point.32 ,49 ,50 Extrafollicular PD-1 expression has been shown to occur in a number of viral lymphadenitides and related conditions, including those involving EBV (figure 2D) and HIV.49 PD-1 positive T-lymphocytes are also an integral component of a number B cell lymphomas, such as nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL).41 Scattered interfollicular CD10-positive lymphocytes should be correlated with terminal deoxynucleotidyl transferase expression as, if positive, they may represent circulating haematogones.45
An entity closely related to AITL is ‘follicular T cell lymphoma’,36 ,37 ,51 where the neoplastic T cells primarily involve follicles, producing appearances resembling progressive transformation of germinal centre (PTGC). There may be admixed CD30, CD15 and EBER-ISH-positive large B cells, and when seen in a PTGC-like background, this lymphoma can resemble lymphocyte-rich (LR) CHL.36 ,37 The important histological clue to this diagnosis is the ‘moth-eaten’ appearances of these follicles due to clusters of atypical T cells expressing TFH markers.36 ,37 The list of diagnostic points and pitfalls is summarised in Table 4.
Neoplastic B cells: nodular lymphocyte-predominant Hodgkin lymphoma
NLPHL constitutes about 5% of all Hodgkin lymphoma52 ,53 and is distinct from CHL in its clinical presentation, behaviour, morphology and immunophenotype.52 NLPHL patients are usually males who present around a decade later than the peak for CHL, that is, 30–40 years old, typically with localised peripheral lymph node enlargement. Despite a propensity for recurrence, the prognosis is usually excellent.52 ,54 The large neoplastic cells in NLPHL are the lymphocyte-predominant (LP) cells (figures 5B and 6B) (also known as ‘popcorn’ cells and formerly called L&H cells).52 ,55 The classic morphological description of NLPHL is that of a lymphoid proliferation with multiple large nodules (figure 6A) comprising reactive IgD-positive small B-lymphocytes associated with expanded FDC meshworks (figure 6C, D), within which are scattered large LP cells that show round to polylobated nuclei with small-to-medium-sized nucleoli.3 ,20 ,54 ,56 ,57 CD21 may be more effective than CD23 in highlighting the FDC meshworks in NLPHL.54 Remnant reactive follicles, if present, are usually seen at the periphery of the involved lymph node and rarely within the lesional nodules.20 ,55 ,56 LP cells morphologically resembling HRS cells may be present, but are usually in the minority, and scattered clusters of epithelioid histiocytes are frequently seen (figure 6A).20 ,55 The LP cells typically show uniform strong expression of B cell markers and transcription factors CD20 (figure 5D), PAX5, Oct-2 (figure 5E) and BOB.1, while CD79a is usually weakly expressed (figure 5E).52 ,57 ,58 BCL6 is also frequently positive (figure 5F) and of higher intensity than MUM1 in the LP cells, consistent with their germinal centre origin, but CD10 is negative.54 ,57 The LP cells may also express epithelial membrane antigen (EMA) and IgD (figure 5F), the latter most commonly in young males.57 ,59 The B cell-rich nodules also contain numerous PD-1-positive TFH cells rosetting the LP cells (figure 5G).60 ,61 LP cells are further distinguished from true HRS cells of CHL by their positivity for CD45/LCA (figure 5F) and negativity for CD30, CD15 and EBER-ISH,20 ,52 ,55 although rare cases expressing these markers exist (CD30 in particular almost always being weak where present).20 ,36 ,52 ,54 ,62–64 A more common scenario is that of accompanying non-neoplastic bystander perifollicular activated cells/immunoblasts that are strongly CD30 positive and occasionally EBER-ISH positive, but are usually smaller than the LP cells.3 ,20 Care must therefore be taken to identify the specific cells positive for CD30 to avoid misdiagnosing NLPHL as CHL (figure 5H). This may be challenging since negatively stained LP cells may be difficult to visualise if the counterstain is weak. In our experience, Oct-2 is the most sensitive reference marker for localising LP cells to compare with other immunostained sections. In recurrences, it should be noted that patients may have had prior treatment with anti-CD20 therapy, so that the LP cells may be CD20 negative.52 ,65
Nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) and T cell/histiocyte-rich large B cell lymphoma

Nodular lymphocyte predominant Hodgkin lymphoma with a predominant T cell-rich diffuse pattern (pattern E): low-power view showing a diffuse infiltrate (A, left, H&E 40×) with abundant CD3-positive T cells rimming scattered lymphocyte-predominant (LP) cells (arrow) (A, right, CD3 100×). LP cells (B, H&E 600×) are round to polylobated, but distinctly larger than the background small lymphocytes. There is only a focal nodule with follicular dendritic cell meshwork (arrow) (C, CD21 40× with inset 200×, open arrow head: LP cell), which is rich in B cells (D, left, CD20 (nodular) 100×). LP cells are strongly positive for CD20 and in the diffuse area there is a paucity of background small B cells (D, right, CD20 (diffuse) 400×). LP cells are also positive for Oct-2 (strong) (E, 400×), CD79a (weak to moderate) (arrow) (E, 400×), BCL6 (arrow), EMA, IgD and CD45/LCA (F, 600×). Rosettes of PD-1-positive follicular helper T cells surrounding the LP cells (arrow) are prominent in the nodular areas (G, left, 600×), but less conspicuous in the diffuse areas (G, right, 600×). There are relatively frequent scattered CD30-positive cells (H, 100×), but these positive cells are non-neoplastic bystander cells (open arrow head) and the LP cells are negative (arrow) (H, inset, 600×). EMA, epithelial membrane antigen; LCA, leucocyte common antigen.

Nodular lymphocyte-predominant Hodgkin lymphoma, typical nodular/serpinginous pattern. This is an unusual recurrence (presenting as diffuse T cell-rich variant but relapsing as nodular typical pattern) from the same case presented in figure 5 following anti-CD20 therapy. Low-power view shows a vaguely nodular architecture (A, H&E 40×), with scattered clusters of epithelioid histiocytes (A, inset, H&E 400×). Lymphocyte-predominant (LP) cells are round to polylobated (arrows) (B, left, H&E 600×) and are negative for CD20 but positive for CD79a (arrows) (B, right, 600×). The nodular and serpinginous patterns are best highlighted by CD21 staining for follicular dendritic cell meshworks (C, 40×), showing follicles with rounded and well demarcated contours. PD-1-positive cells are numerous within the nodules (D, left, 40×). The nodules are also rich in IgD-positive small B-lymphocytes (D, right, 100×) and, as previously, the LP cells are IgD positive (arrow).
In the context of a T cell-rich infiltrate with large B cells, the variant patterns of NLPHL deserve special mention. Fan et al56 described six morphological patterns of NLPHL, namely the ‘classic’ B cell-rich nodular (pattern A, described above); serpinginous/interconnected (pattern B, a variation of pattern A with distinct FDC meshworks within serpinginous nodules); prominent extranodular LP cells (pattern C); T cell-rich nodular (pattern D, with FDC meshworks in the nodules but reduced small B cells); diffuse T cell-rich pattern (pattern E, with loss of FDC meshworks and a paucity of small B cells); and diffuse ‘moth-eaten’ B cell-rich pattern (pattern F, usually with FDC meshworks but lacking nodules). Patterns A and B are considered the ‘typical’ pattern, while patterns C to E are considered ‘variant’ patterns.52 ,53 Pattern F is very uncommon. A mixture of patterns may be seen.56 Variant patterns may be associated with more advanced disease, more frequent relapses and poorer overall prognosis, and should be described in the histopathology report.39 ,53 ,56 The T cell-rich patterns present a particular diagnostic challenge, especially when these are diffuse (pattern E) and predominant (figure 5A, C), since there is a morphological overlap with T cell/histiocyte-rich large B cell lymphoma (TCHRLBCL), such that this diffuse pattern is often called ‘TCHRLBCL-like’.56 By definition, de novo TCHRLBCL can be excluded by even the focal presence of a nodule of more typical NLPHL pattern (figure 5C), emphasising the need for generous sampling and performing immunohistochemistry for B cells and FDC meshworks on multiple sections.54 ,62 The presence of numerous admixed small B cells is also highly unusual for TCHRLBCL.54 IgD positivity in the LP cells tends to favour NLPHL over TCHRLBCL, which is usually negative for IgD.59 ,62 PD-1-positive T cell rosettes may be present in both conditions or may be scanty in diffuse areas of NLPHL (figure 5G).61 Recurrences in NLPHL may sometimes show evolution from the more typical patterns to diffuse T cell-rich patterns, although the reverse may infrequently occur (figures 5 and 6).56 Bone marrow involvement by NLPHL is usually T cell-rich in pattern irrespective of the pattern in the accompanying lymph node.54 ,66 In summary, in small core biopsies and bone marrow trephines, the pathologist should exercise caution when considering a diagnosis of de novo TCHRLBCL, and clinical and radiological correlation is important as the TCHRLBCL frequently presents in a more aggressive manner than NLPHL.54 ,55 The distinction between NLPHL with diffuse T cell-rich pattern and TCHRLBCL may not be possible on core biopsies and in these cases must be deferred to an excision lymph node biopsy.55
The presence of numerous PD-1-positive T cells accompanied by scattered large B cells, expanded and/or irregular appearing FDC meshworks, and sometimes CD30 and EBER-ISH-positive bystander cells in NLPHL may raise the possibility of AITL. Such resemblance is normally superficial however as the pattern of FDC meshworks is usually distinctive. At low power, FDC meshworks in NLPHL tend to be more rounded and well circumscribed (figure 6C), in contrast to AITL (patterns 2 and 3), which shows spiky irregular FDC meshworks with tentacular extension encircling HEVs (figure 4B). In addition, while eosinophils and plasma cells are rarely seen in NLPHL,20 these cells are frequent in AITL, which also shows atypical T cells. Some cases of NLPHL, especially in younger patients, may show activated and morphologically mildly atypical T cells admixed with LP cells expanding the paracortex of a partially involved lymph node, raising the possibility of PTCL, but in these cases, typical NLPHL areas are usually also present and there is neither immunophenotypic nor molecular genetic evidence of PTCL.67 The list of diagnostic points and pitfalls is summarised in Table 5.
Neoplastic B cells: T cell/histiocyte-rich large B cell lymphoma
TCHRLBCL is a variant of DLBCL characterised by a minority (under 10%) of neoplastic large B cells scattered in a reactive T cell-rich background, usually with admixed non-epithelioid histiocytes (figure 7A, B).68–70 There is a slight male preponderance and patients are usually middle-aged or older adults. The disease usually presents in an advanced stage and has a poor prognosis.54 ,69 ,71 The neoplastic large B cells are singly scattered without aggregation and may assume a range of morphologies, including LP cell-like, HRS-like or immunoblast/centroblast-like.68 ,69 These large B cells immunohistochemically express CD45/LCA and the full range of B cell markers and transcription factors (ie, CD20, CD79a, PAX5, Oct-2, BOB.1) (figure 7C, D).52 ,68 They are commonly positive for BCL6, sometimes positive for EMA (figure 7D), and usually negative for CD30 and CD15.68 ,69 ,72 This immunohistochemical profile helps to distinguish TCHRLBCL from CHL, but not NLPHL, in which the LP cells share a similar immunophenotypic profile.68 IgD expression by the large B cells is distinctly rare in de novo TCHRLBCL, unlike NLPHL.59 ,62 By definition, EBV should be negative as morphologically similar lesions with EBV-positive neoplastic large B cells are classified as EBV-positive DLBCL.62 ,68 ,71

T-cell/histiocyte-rich large B cell lymphoma: pleomorphic cells with immunoblastic and Hodgkin/Reed-Sternberg-like features (arrows) and background of reactive inflammatory cells (A, H&E 400×), comprising numerous CD3-positive T cells (B, left, 400×) and CD163-positive histiocytes (B, right, 400×). The scattered large cells are negative (arrow) for CD3 and CD163 (B, CD3 and CD163, 400×) and are B cells highlighted by CD20 (C, 400×). The large B cells are positive for CD79a, Oct-2, BCL6 and EMA (D, 600×). EMA, epithelial membrane antigen.
The background reactive T cell infiltrate in TCHRLBCL is usually dominated by CD8-positive non-activated cytotoxic T cells (ie, TIA-1 positive, granzyme B negative).68 ,73 FDC meshworks are absent and reactive small B-lymphocytes should be absent or scarce.69 ,72 ,74 While this microenvironment may distinguish TCHRLBCL from typical NLPHL, it cannot reliably differentiate it from the T cell-rich diffuse variant pattern of NLPHL, especially in small biopsies. As in NLPHL, rosetting of large B cells by PD-1-positive T cells may sometimes be seen in TCHRLBCL.61 As previously noted, close clinical correlation is important. There is in fact substantial genetic overlap between LP cells in NLPHL and neoplastic B cells in TCHRLBCL, although it remains controversial if TCHRLBCL represents a biological progression from NLPHL.75 ,76 If a patient has a history of NLPHL, but has a synchronous or metachronous lesion that is entirely diffuse and meets morphological criteria for TCHRLBCL, the current recommendation is to call it ‘TCHRLBCL-like transformation of NLPHL’.39
Rare cases of TCHRLBCL have been described that show atypical appearing T cells positive for PD-1 and showing reduced CD7 expression, raising the possibility of TFH-type PTCL (eg, AITL). However, these showed no prominent infiltrate of eosinophils or plasma cells nor increased vascularity. CD10 was not expressed in the T cells and other T cell antigens were preserved, while no clonal rearrangements were detected on T cell clonality studies.70 ,77 TCHRLBCL should show clonal immunoglobulin gene rearrangement since the B cell population is almost entirely neoplastic.65 ,74 The list of diagnostic points and pitfalls is summarised in Table 5.
Neoplastic B cells: classical Hodgkin lymphoma
CHL is one of the most common lymphoma subtypes to show a minority of large B cells in a T cell-rich background. Its morphological features are well known, that is, large mononuclear Hodgkin cells and binucleated/multinucleated Reed-Sternberg cells in an appropriate non-neoplastic inflammatory background comprising small to occasionally medium-sized lymphocytes, histiocytes, eosinophils and plasma cells.20 ,54 HRS cells contain large vesicular nuclei with eosinophilic ‘viral inclusion-like’ nucleoli and are usually scattered, although in some cases may show prominent clustering. There are four main subtypes of CHL, namely nodular sclerosis (NS), mixed cellularity (MC), LR and lymphocyte-depleted (LD). NS-CHL and MC-CHL are the most common.3 HRS cells are B cells strongly positive for CD30 and MUM1, usually weak to moderately positive for PAX5 and may also be positive for CD15 and/or EBV by EBER-ISH and latent membrane protein (LMP)-1. They are negative for CD45/LCA, ALK-1 and T cell markers.20 ,78 Uniform strong expression of commonly used B cell markers (CD20 and CD79a) and transcription factors (Oct-2 and BOB.1) is not seen in HRS cells but focal/heterogeneous and weak expression of these markers is not uncommon.58 ,79 ,80 Hence, the assessment of B cell markers and transcription factor expression by immunohistochemistry in CHL should be based on the overall pattern across the HRS cell population as a whole rather than that in individual cells. A lymphoma showing morphological features of CHL with strong CD30 expression in the HRS cells, but with uniform strong expression of B cell markers and transcription factors, is best classified as ‘B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL’.62 In our experience, MUM1 is very sensitive (though not specific) for HRS cells (figure 8D) and is useful for localising HRS cells for comparison with other immunohistochemical markers.81
Classical Hodgkin lymphoma (CHL)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
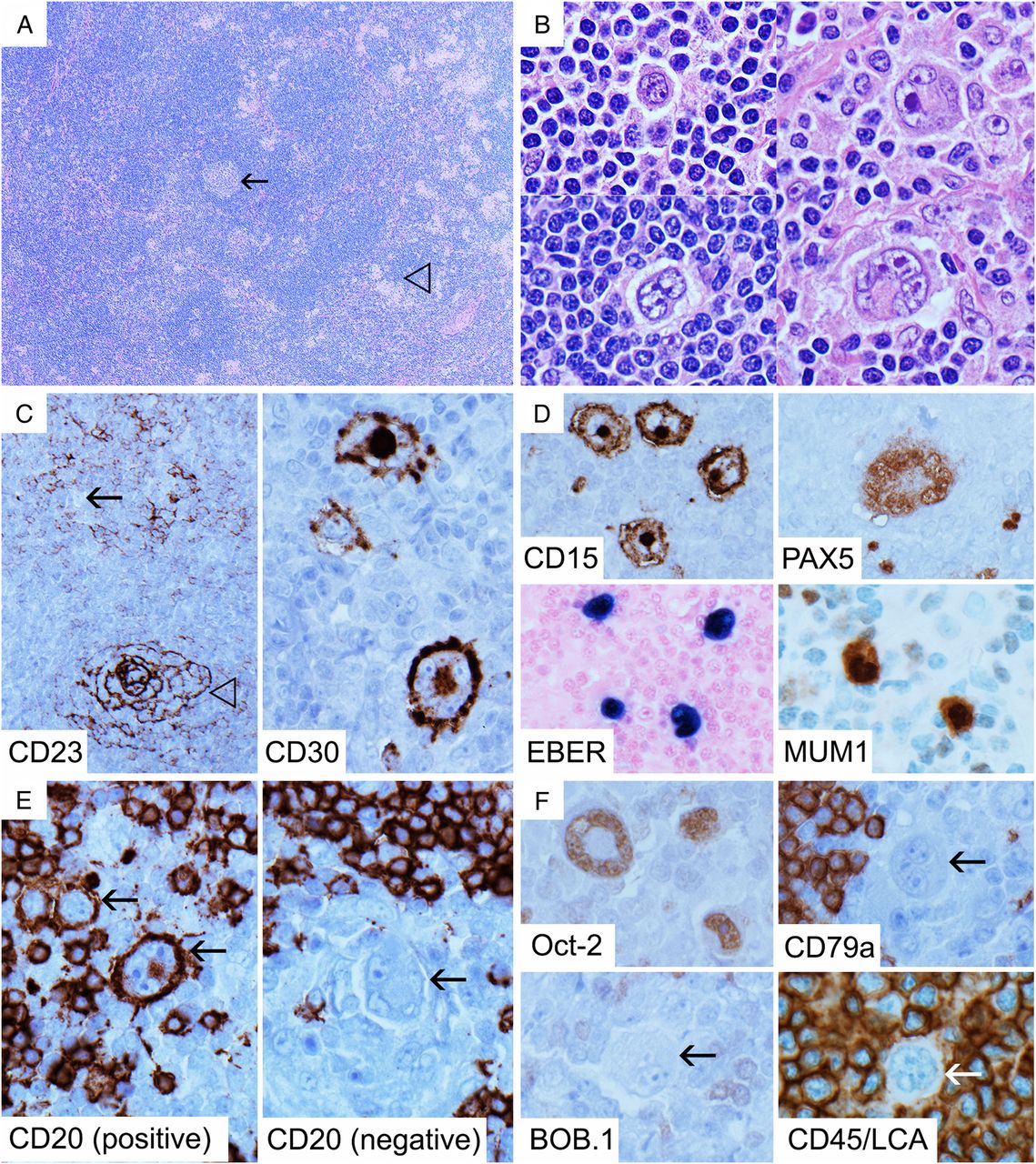
Lymphocyte-rich classical Hodgkin lymphoma: low power shows a nodular appearance with remnant germinal centres (arrow) and clusters of pale epithelioid histiocytes (open arrow head) (A, H&E 40×). Hodgkin/Reed-Sternberg (HRS) cells show various morphologies, ranging from lymphocyte-predominant cell-like (B, left, H&E 600×) to typical Reed-Sternberg cells (B, right, H&E 600×). CD23 shows an atrophic germinal centre highlighted by strong concentric staining for follicular dendritic cell (FDC) meshworks (open arrow head), with expanded mantle zone showing weakly staining FDC and small B cells containing negative HRS cells (arrow) (C, left, 100×). HRS cells are strongly positive for CD30 (C, right, 600×), CD15, MUM1 and Epstein-Barr virus by in situ hybridisation for EBER (EBER-ISH) (D, 600×). PAX5 shows weak-to-moderate expression (D, 600×). CD20 is positive in some HRS cells (arrow) but negative in others (E, 600×), and although Oct-2 is weak to moderately expressed, HRS cells (arrow) are negative for CD79a, BOB.1 and CD45/LCA (F, 600×). LCA, leucocyte common antigen.
There are many histological mimics of CHL (see preceding sections), but its most consistent defining feature is an appropriate background cellular milieu. In contrast to its reactive mimics, the presence of an prominent immunoblastic response with a spectrum of plasmacytic differentiation is atypical for CHL, although mature plasma cells may be present.82 In the context of its neoplastic mimics, areas of otherwise typical non-Hodgkin's lymphoma (eg, AITL) intermingled with cells that would otherwise fit the morphology and immunophenotype of HRS cells are inconsistent with CHL. Composite CHL and non-Hodgkin's lymphoma should only be diagnosed if there are separate clear-cut areas consisting exclusively of each entity.33 ,36 Purely extranodal involvement of CHL without lymph node involvement is also very rare20 and localised mucosal ulcerative lesions resembling EBV-positive CHL are more likely to represent the recently described indolent entity ‘EBV-positive mucocutaneous ulcer’ that may be age/senescence or immunosuppression related.39 ,83
We end this section with a brief description of LR-CHL, described in the latest revised WHO classification as having characteristics intermediate between NLPHL and CHL.39 This relatively uncommon subtype of CHL is generally associated with early stage disease and a relatively good prognosis, and tends to occur in adult males.54 ,84 LR-CHL often has a nodular architecture (figure 8A), without the accompanying fibrous sclerosis that defines NS-CHL, and may morphologically mimic NLPHL.84 The nodules usually comprise eccentric regressed germinal centres with expanded mantle zones (figure 8C), and HRS cells are present in the mantle zones as well as between the follicles.20 ,54 As in NLPHL, clusters of epithelioid histiocytes may be seen and eosinophils are rare, though as compared with NLPHL, the nodules are often smaller and remnant germinal centres are more common (figure 8A).20 ,55 HRS cells may have classical appearances as described above or may resemble LP cells (figure 8B). Furthermore, HRS cells of LR-CHL more commonly express B cell markers and transcription factors (figure 8E, F), and may be surrounded by rosettes of PD-1-positive small T cells, further simulating NLPHL.85 Hence, demonstration of strong CD30 (figure 8C) positivity in HRS cells with or without CD15 and/or EBER-ISH positivity (figure 8D), as well as negativity for CD45/LCA (figure 8F), is important in distinguishing LR-CHL from NLPHL. The list of diagnostic points and pitfalls is summarised in Table 6.
Conclusion
We have provided an overview and discussion of the main entities that the pathologist may encounter manifesting as a lymphoid proliferation composed predominantly of small T cells together with smaller numbers of large B cells. Although it is easy to focus on the most atypical-appearing element of the infiltrate in this group of conditions, many of the most important diagnostic clues lie in architectural features and the background cellular milieu. Careful assessment of the architecture and the entire cellular population including FDCs and inflammatory cells is therefore the most important general principle in achieving the correct diagnosis. A suggested diagnostic approach is presented in table 7.
Diagnostic approach and advice for assessing lymphoid lesions with a T cell-rich infiltrate and minor large B cell component
Take home messages
Assessment of a lymphoid population composed predominantly of small T cells, together with smaller numbers of large B cells, is a common diagnostic problem. Accurate diagnosis of such an infiltrate requires close evaluation of the overall lymphoid architecture and the inter-relationships between B-cells and T cells, follicular dendritic cells and sometimes other inflammatory cells.
An integrated approach, with close correlation of patient age, past and current medical and treatment history and, clinical presentation, morphology and, where applicable, molecular clonality studies, is required to avoid pitfalls and misdiagnosis.
Beware of the limitations of small biopsies, especially where the lymphoid architecture cannot be assessed with confidence. If uncertainty remains regarding definitive diagnosis, it is best to err on the side of caution and consider obtaining a larger and more representative specimen.
References
Footnotes
Handling editor Cheok Soon Lee
Contributors CLC wrote the manuscript. SO provided guidance and significant input for refinement of the work.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.