Article Text

Abstract
Aims A standard approach in test evaluation is to compare results of the assay in validation to results from previously validated methods. For quantitative molecular diagnostic assays, comparison of test values is often performed using simple linear regression and the coefficient of determination (R2), using R2 as the primary metric of assay agreement. However, the use of R2 alone does not adequately quantify constant or proportional errors required for optimal test evaluation. More extensive statistical approaches, such as Bland-Altman and expanded interpretation of linear regression methods, can be used to more thoroughly compare data from quantitative molecular assays.
Methods We present the application of Bland-Altman and linear regression statistical methods to evaluate quantitative outputs from next-generation sequencing assays (NGS). NGS-derived data sets from assay validation experiments were used to demonstrate the utility of the statistical methods.
Results Both Bland-Altman and linear regression were able to detect the presence and magnitude of constant and proportional error in quantitative values of NGS data. Deming linear regression was used in the context of assay comparison studies, while simple linear regression was used to analyse serial dilution data. Bland-Altman statistical approach was also adapted to quantify assay accuracy, including constant and proportional errors, and precision where theoretical and empirical values were known.
Conclusions The complementary application of the statistical methods described in this manuscript enables more extensive evaluation of performance characteristics of quantitative molecular assays, prior to implementation in the clinical molecular laboratory.
- molecular pathology
- cancer genetics
- diagnostics
- statistics
- quantitation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The use of appropriately stringent statistical methods in assessment and comparison of clinical molecular laboratory assays during validation is crucial to ensure appropriate test interpretation and avoid erroneous conclusions.1 With the recent uptake of next-generation sequencing (NGS) in clinical molecular diagnostic labs, and increased use of quantitative values from NGS in clinical tests, there is a need to improve statistical methodologies used by clinical molecular laboratories for assessment and comparison of quantitative assays. Historically, clinical molecular diagnostic laboratory tests often yielded binary results, such as presence or absence of specific gene variants.1 Implementation of NGS and other novel techniques in clinical laboratories enabled more uses of quantitative data, such as in determination of variant allele frequency (VAF; ie, values for reference and alternative nucleotides at specific genomic positions) in tumour profiling, or gene expression levels (see, eg,2–5). VAF is useful in examining such characteristics as tumour heterogeneity, clonal architecture and tumour evolution.6 Furthermore, VAF is associated with clinical outcome for leukaemia patients with FMS-like tyrosine kinase-3 (FLT3) internal tandem duplications.7 Therefore, accurate quantitative measurements of VAFs would be useful for patient management and care. Although other laboratory specialties (eg, clinical chemistry) that have a more fundamental reliance on quantitative assays have used more extensive statistical methods for quantitative assay comparison studies,8 molecular diagnostic laboratories have not widely adopted these methods. Therefore, there is a need to improve the statistical approaches used to evaluate quantitative molecular methods, including NGS and bioinformatic approaches, to more fully understand the limitations of NGS assays in the clinical laboratory.
Currently, use of simple linear regression (SLR) and the coefficient of determination (R2) is commonplace in manuscripts assessing quantitative aspects of NGS platform comparison,9 NGS assay development and validation studies10 11 and in comparison studies of quantitative non-NGS molecular assays such as quantitative PCR and digital droplet PCR.4 12 13 R2 represents the proportion of variation explained by a given model and is typically used as a substitute for degree of agreement, or how closely measurements from two quantitative assays agree with each other. Although R2 can be used as an indicator of correlation, it does not adequately assess constant and proportional errors. Constant error (‘bias’ in the context of Bland-Altman (BA) analysis14) is the difference between two methods, or a method and a reference, that does not change over a given reportable range. Proportional error (‘systematic error’) is the difference between two methods, or a method and a reference, that changes over a given reportable range.15
The BA method was initially developed to address the shortcomings of SLR and R2 for evaluation of laboratory tests.14 16 BA analysis recommends the following minimum components for making conclusions on agreement: bias and limits of agreement; definition of acceptable agreement; precision of the estimated limits of agreement; relationship between difference and magnitude; and the importance of repeatability.17 BA analysis yields a plot useful in qualitatively assessing differences in measurements between two assays (figure 1). In addition to visual examination of the data, BA analysis includes statistical tests to determine the presence and magnitudes of constant error, proportional error and the degree of agreement (see online supplementary materials and methods). The values may also be plotted as a percentage in situations with high variability.18 Notably, BA has been widely used by the diagnostic community to evaluate clinical laboratory tests for several decades.14 16 The method has been cited over 30 000 times in the literature, and certain clinical journals require authors to report the results of BA as part of all assay comparison studies.19 Despite ubiquitous use of BA in other clinical diagnostics areas,19 it is seldom used in evaluating molecular diagnostic assays, where SLR usage is prevalent.

Interpretation of Bland-Altman (A) and Deming/simple linear regression (B) plots for the purpose of assay comparison. Black dashed lines display the distribution of differences in measurements by the two assays in a Bland-Altman plot and are used to estimate the degree of agreement (A). Constant error (solid blue line), proportional error (red dashed line) and 95% limits of agreement (black dashed line) are displayed in Bland-Altman plot (A). In a correlation plot, the slope of the red dashed line is compared with 1 (shown by the black solid line indicating perfect agreement) (B). Although constant error is not directly visualised in a correlation plot, the position of the red dashed line in relation to the black solid line (perfect agreement) may be used as a surrogate (A). In a Bland-Altman plot, the blue line indicates constant error or the average difference in measurements by the two assays (B). Precision of an assay may be estimated using degree of agreement (A) or prediction intervals (B) (black dashed lines) for spike and recovery experiments.
In this manuscript, we examined the utility of BA and the enhanced linear regression analysis methods, Deming regression (DR) and SLR, for assessment and comparison of NGS data, including VAF measurements and bioinformatics tool outputs, using data sets from targeted NGS panels for somatic tumour profiling. Furthermore, we present the adaptation of BA and SLR to measure precision and accuracy of NGS data and superiority of these methods for assessing test performance. The complementary application of BA, DR and SLR enables more comprehensive assessment of quantitative assay performance for NGS than relying on R2 alone. Use of BA, DR and SLR should be considered by molecular diagnostic laboratories validating NGS for quantitative measurements in order to obtain key assay performance metrics during quantitative assay comparison and validation.
Methods
Statistical analysis
Statistical analyses were performed in R using custom scripts,20 provided in online supplementary file S1. BA plots were generated using a modified ‘baplot.R’ script (https://gist.github.com/jmmateoshggm/5599056; last accessed 13 October 2016), DR was performed using a modified ‘mcDeming.r’ script (https://CRAN.R-project.org/package=mcr; last accessed 13 October 2016). Custom R scripts were validated against results from EP Evaluator Software (Data Innovations, Burlington, Vermont, USA). BA analysis was performed as previously described14; constant error was determined by comparing the mean of differences measurements using a one-sample t-test ( ). The slope of the line of best fit was used to estimate proportional error for BA, using the average signal as the explanatory variable and the difference between signals as the response variable. Degree of agreement in BA was determined by the distribution of values of the differences in measurements (95% CI). In the context of BA, precision was estimated using degree of agreement while using theoretical values as a variable.
). The slope of the line of best fit was used to estimate proportional error for BA, using the average signal as the explanatory variable and the difference between signals as the response variable. Degree of agreement in BA was determined by the distribution of values of the differences in measurements (95% CI). In the context of BA, precision was estimated using degree of agreement while using theoretical values as a variable.
For linear regression methods, the difference in means between the measurements of two assays was used to quantify constant error, which was tested using a paired t-test ( ). The slope of the line of best fit was tested to determine presence and magnitude of proportional error (
). The slope of the line of best fit was tested to determine presence and magnitude of proportional error ( ). Prediction interval (95%) range was used to estimate precision for spike-and-recovery data.
). Prediction interval (95%) range was used to estimate precision for spike-and-recovery data.
Data sets
To evaluate the statistical methods of BA, DR and SLR, the following data sets were used. (1) VAFs of 486 variants identified from NGS testing of 349 DNA samples extracted from formalin-fixed, paraffin-embedded (FFPE) tumour tissues from variant solid tumours using a targeted NGS panel (TruSeq Amplicon Cancer Panel; Illumina, San Diego, California, USA) on the MiSeq (Illumina) bench top sequencer, using methods previously described21; (2) VAFs from Sanger sequencing of the same 486 variants from 349 DNA samples, described in (1). Sanger sequencing was performed using a custom library of primers covering variants of interest amplified using the ProFlex PCR System (Thermo Fisher Scientific) and sequenced on 3500XL Genetic Analyzer (Thermo Fisher Scientific) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). (3) A subset of variants (n=166, primarily consisting of hotspot variants in KRAS, PIK3CA and TP53) recurrent in tumour tissue from the 349 samples selected for comparison of bioinformatics tools BWA/MuTect22 23 (settings: -dt None --max_alt_allele_in_normal_fraction 0.03 --max_alt_alleles_in_normal_count 1000000 --max_alt_alleles_in_normal_qscore_sum 1000000 --gap_events_threshold 1000000 --pir_median_threshold 1) and NextGENe using previously described settings21 (SoftGenetics, State College, Pennsylvania, USA). (4) Variant data from a previously published cell line dilution experiment,10 used for precision and accuracy calculations. Within our lab, precision and accuracy are defined as previously described.24
Results
Validation of BA and linear regression scripts using EP Evaluator
To validate the scripts used to analyse method comparison data in R, commercially available software (EP Evaluator) was used. For each dataset, EP Evaluator was used on the identical measurements to generate a single report for each method comparison data set (see online supplementary file S2). EP Evaluator reports include both BA and scatter plots analysed by both SLR and DR approaches. R and EP Evaluator were concordant with respect to regression line equations (slope and y-intercept), degree of agreement, constant and proportional errors ensuring that the custom R scripts were validated to work as intended for dataset evaluations.
VAF comparison and evaluation
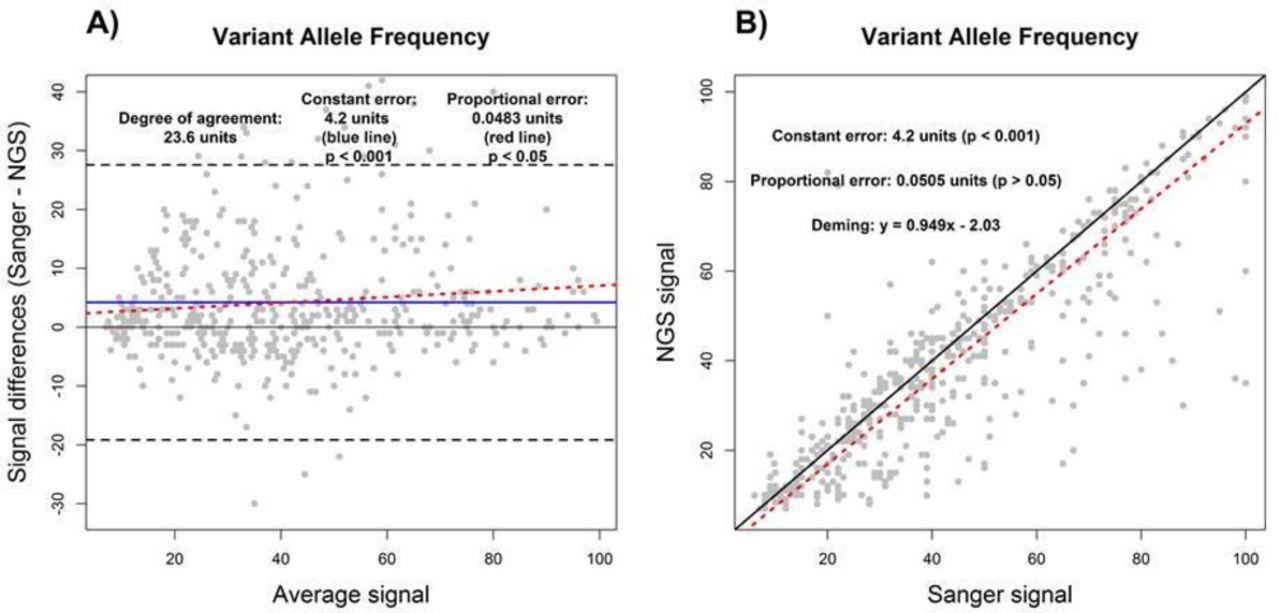
We first sought to demonstrate the utility of BA and DR by assessing measures of constant and proportional error in VAF measurements from NGS and Sanger sequencing tests on a set of 349 FFPE tumour samples, as described in the Methods. Using R2 derived from SLR to compare the VAF measurements from NGS and Sanger showed an R2 value of 0.76, indicating a less than perfect agreement between NGS and Sanger results for measurement of VAF (data not shown). Although the sources of disagreement could not be ascertained by use of R2 alone, both DR and BA agreed on the presence of constant error (figure 2). As shown by the constant error values in figure 2A and B, Sanger sequencing measurements of VAF were higher than NGS VAF by an average of 4.2% (p<0.001) across the reportable range. In addition, a proportional error of 0.0483% (p<0.05) was detected by BA analysis, while the value of 0.0505% identified by DR was not statistically significant (p>0.05). This indicates that the differences in VAF measurements made by NGS and Sanger sequencing may be greater at the high end of the reportable range depending on the analysis method used (figure 2; also see per cent bias plot in online supplementary file 2). Lastly, the use of BA and DR indicates that VAF measurements between NGS and Sanger sequencing may differ by up to 23.6% according to the degree of agreement determined by BA analysis (figure 2A).
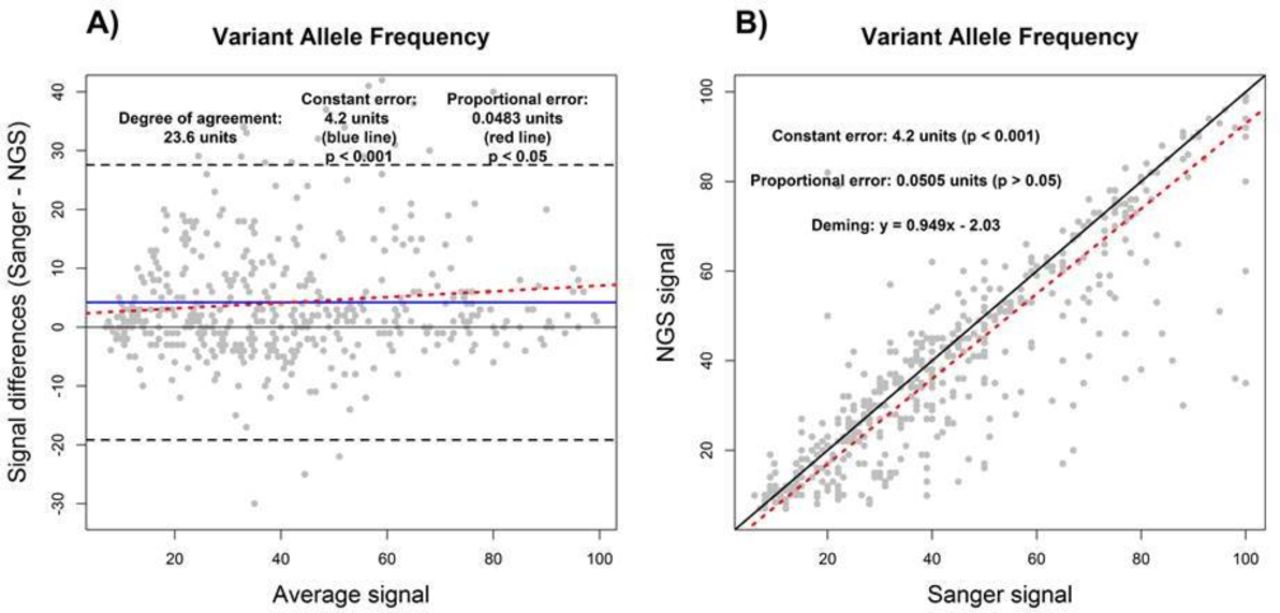
Assay comparison analysis using the Bland-Altman (A) and Deming linear regression methods (B). Variant allele frequency results as measured by NGS were compared with Sanger sequencing. Constant error (solid blue line), proportional error (red dashed line) and degree of agreement (black dashed line) are displayed in Bland-Altman plot. For correlation plot, perfect correlation (ie, slope of 1) (black solid line) and the Deming linear regression (red dashed line) are displayed. The degree of agreement is estimated by the prediction intervals (black dashed lines). The presence and magnitudes of performance characteristics are displayed for both Bland-Altman and correlation plots. NGS, next-generation sequencing.
Bioinformatics tool comparison and evaluation
Quantitative outputs of bioinformatics tools used to analyse NGS data can also be evaluated by BA and DR. For the purpose of this study, NGS coverage of a subset of 166 variants (primarily recurrent hot spot codons: eg, KRAS G12/13, TP53 R175 and PIK3CA H1047) detected by the NGS tumour panel (measured as the number of reads that cover a given genomic position) and VAF were compared between BWA-MuTect and NextGENe. Of note, NextGENe counts all overlapping reads from paired end sequenced amplicons, unlike BWA-MuTect that does not, thus introducing a known source of constant and proportional error between these two tools.
For VAF, there was no constant error detected between MuTect and NextGENe by both approaches; however, the discrepancies in proportional error were once again evident between BA and DR (figure 3A and B). By itself, the R2 value of 0.98 for the VAF measurements determined by SLR (data not shown) indicated a high degree of agreement between MuTect and NextGENe, correlating with the results found by BA and DR. However, the R2 value of 0.93 determined using SLR (data not shown) for read depth coverage between MuTect and NextGENe outputs would be interpreted as a measure of high degree of agreement. However, as evident in our BA and DR evaluations, read depth coverage as calculated by MuTect was on average 1950× lower than NextGENe, as shown by the presence of a constant error of 1950 units (figure 3C and D). In addition, MuTect read depth coverage values were also 0.59 and 0.54 that of NextGENe as determined by BA and DR, respectively, indicating the presence of proportional error with higher read depth from NextGENe analysis (figure 3C and D; also see percent bias plot in online supplementary file S2). Lastly, the degree of agreement was estimated to be 2920× by BA (figure 3C). In this data set, R2 alone was not a sufficiently adequate method to compare these tools with respect to read depth coverage due to the presence of significant constant and proportional error undetectable by use of R2 alone.
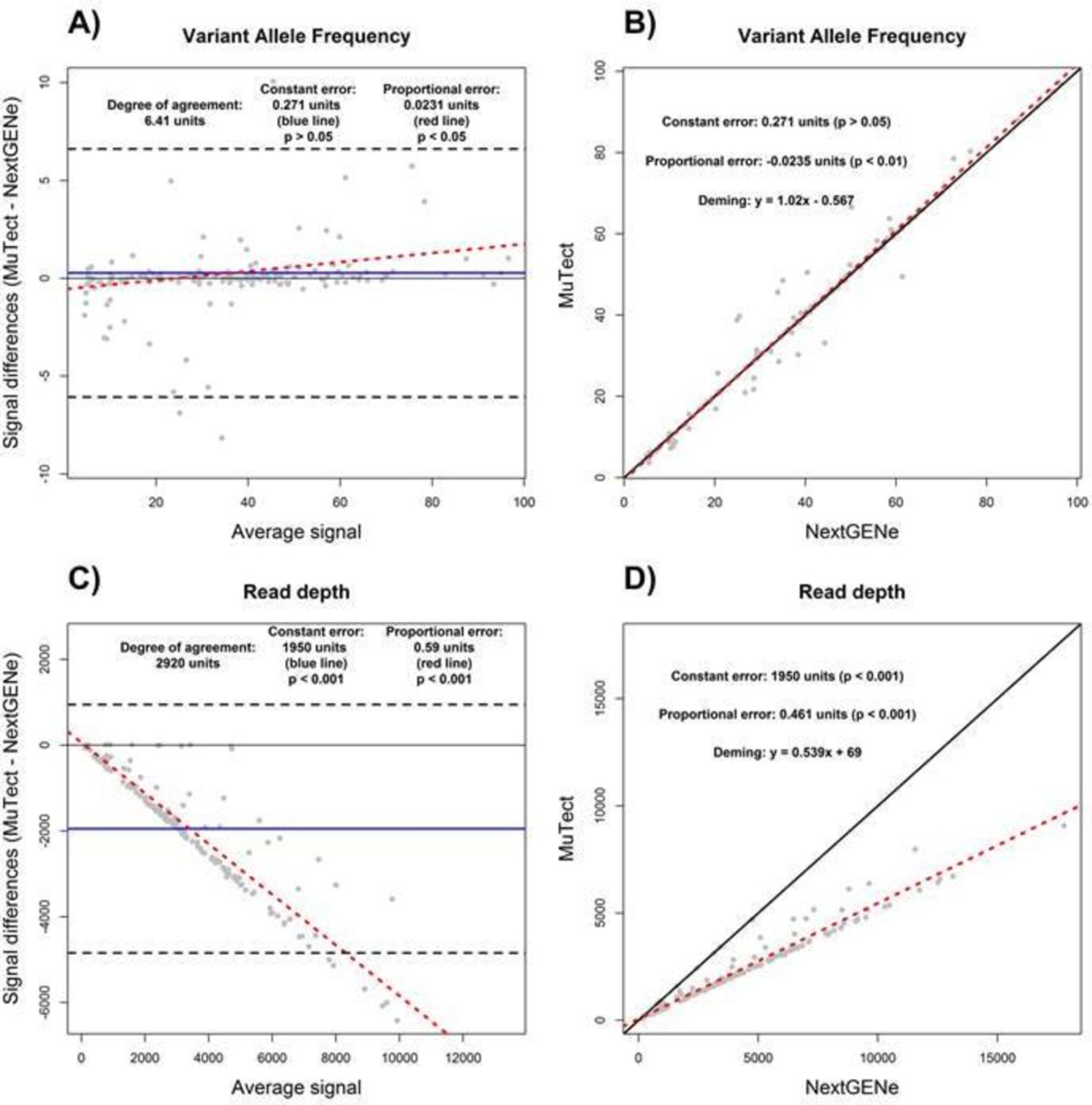
Bioinformatics tools comparison analysis using the Bland-Altman (A and C) and Deming linear regression methods (B and D). The variant allele frequency measurements (A and B) and read depth coverage (C and D) as determined by BWA-MuTect and NextGENe are displayed. Constant error (solid blue line), proportional error (red dashed line) and degree of agreement (black dashed line) are displayed in Bland-Altman plot. For correlation plot, perfect correlation (ie, slope of 1) (black solid line) and the Deming linear regression (red dashed line) are displayed. The degree of agreement is estimated by the prediction intervals (black dashed lines). The presence and magnitudes of performance characteristics are displayed for both Bland-Altman and correlation plots.
Precision and accuracy assessment
Precision and accuracy are required metrics for evaluating quantitative assays as specified in clinical laboratory standards.25 The assessment of these characteristics can be accomplished using spike-and-recovery (eg, cell line dilution) experiments that challenge the assay with a known amount of the analyte of interest over a range of values for quantitative assays. Accuracy (determinate error) represents the closeness of agreement between measurements and the true value and is composed of both proportional and constant error.26 Precision (indeterminate error) is the variability between individual measurements.26 Although BA and SLR have been mostly used for assay comparison, they can also be applied to quantify precision as well as constant and proportional errors of an assay with minimal modifications.
We used a data set from a published validation manuscript,10 which consisted of NGS VAF measurements from a cell line dilution experiment (spike-and-recovery), in order to demonstrate the utility of BA and SLR. A cell line dilution experiment provides both expected (theoretical) and observed (empirical) VAF values, thus enabling quantification of precision and accuracy of an NGS assay. For the published data set, although high correlation between the expected and observed values was noted,10 accuracy and precision were not explicitly quantified. Using BA and SLR, we determined that the precision of the assay was 3.24%, or 3.19%–3.2%, for the single nucleotide variants, and 11.8%, or 11.6%–12.3%, for insertion and deletion variants, for BA and SLR, respectively (figure 4). Both methods were in agreement regarding the presence of constant error of 0.24% and 1.15% for the single nucleotide and insertion/deletion variants, respectively (figure 4). The proportional error as determined by BA analysis was 0.0056% for single nucleotide variants and 0.0073% for insertions/deletions (figure 4). However, the proportional error determined by SLR was 0.99% for single nucleotide variants and 0.93% for insertions/deletions (figure 4). Therefore, both BA and SLR can be used to determine accuracy (ie, constant and proportional errors) and precision for an NGS assay with minor adaptations, although the results may differ depending on the chosen analysis method.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Determination of assay accuracy and precision using Bland-Altman (A and C) and simple linear regression (B and D) methods. The variant allele frequency measurements from a cell line dilution experiment for single nucleotide variants (A and B) and insertions/deletions (C and D) were used to estimate assay accuracy and precision. Degree of agreement (A and C) and prediction intervals (B and D) were used to estimate precision based on expected (theoretical) and observed (empirical) values. Accuracy, including constant (blue line) and proportional (red dashed line) errors, are displayed in both Bland-Altman and correlation plots.
Discussion
We used a number of examples to demonstrate the utility of BA, DR and SLR for the purpose of molecular diagnostic assay comparison and evaluation, with a special focus on application to NGS tests. Since NGS has the potential to supplant many well-established molecular assays, many molecular diagnostic laboratories are evaluating the performance of NGS assays compared with other methods. However, the common use of SLR and R2 in assay comparison studies does not adequately address the determination of constant error, proportional error and degree of agreement, particularly for assays with quantitative measurements such as NGS. Statistical approaches used for assessing assay performance in other diagnostic fields, such as clinical chemistry, are well suited for assay comparison, evaluation and validation in molecular diagnostics, including NGS assays.
Appropriate sample sizes in validation studies are essential to ensuring that the 95% limits of agreement are properly estimated. When considering NGS validation, ‘sample size’ can be considered as the total number of variants detected by the assay. While larger numbers of variants yield more statistical power, this needs to be balanced against practical considerations such as cost of sequencing a larger footprint and sufficient starting template material. Recent analysis in this area has suggested that the minimum number of variants in a validation ought to be 100, with a recommended number estimated at 200, in order to obtain a CI of a maximum of ±0.34 s (where s is SD).27
Also of relevance to the comparison of a new assay such as NGS with an existing lab-standard assay is the contextual difference between ‘intratest’ precision (or intratest repeatability) and ‘intertest’ precision. The former may be determined using samples with known measurement values and including them in multiple repeats of the assay over time. If the intratest precision has not been evaluated, it becomes difficult to perform intertest evaluations to determine which methods being compared are more precise as the agreement between the two methods will likely be poor. One approach to measure intratest precision in NGS assays has been to use well-characterised reference samples with publicly available variant information, for example, the well-characterised HapMap cell line NA12878.
Comprehensive assessment of new methods allows clinical laboratories to stay up-to-date with novel technologies and to continually improve on the diagnostic value of clinical tests. Appropriate statistical analysis of the data from assay comparison studies is essential to demonstrate that a given new test can deliver results that are, at minimum, as reliable as its predecessor. Unfortunately, there have been concerns raised about the quality of assay comparison studies,1 and in particular the statistical analysis of the data. One of the ways to address these concerns is to provide the necessary performance metrics in every assay comparison manuscript and to clearly outline the methodology that was used to obtain these metrics. For example, precision and accuracy, including constant and proportional errors, should be reported for spike-and-recovery experiments, and constant and proportional errors with respect to a reference method should be included in assay comparison studies.
Degree of agreement between two assays in BA analysis serves as a numerical yet qualitative metric to assess whether the two assays are interchangeable28 and should not be used without consideration of clinical judgement to determine whether any two given assays are equivalent for use. Additionally, what constitutes ‘acceptable’ agreement—that is, what minimum level of agreement (or, maximum difference in measurements) in measurement values is required—for two assays to be considered equivalent is a clinical decision, not a statistical one. Significant bias as measured by statistical methods does not yield sufficient information for agreement between two methods to be determined. For example, two methods may have significant difference in bias and yet may agree from clinical point of view and vice versa. Factors that influence this choice may include detection ranges, limits of detection and ranges of clinically significant values, as well as the identities of the variants being measured—for example, a lab will need to consider whether a 10% difference in VAF is acceptable when measuring heterozygous or homozygous germline variants by NGS versus Sanger sequencing and may make a different choice when considering somatic variants at VAF of 10%.
DR is often preferred over SLR in assay comparison studies.29 In general, SLR and DR can include additional steps to calculate constant and proportional errors similar to BA, although ‘enhanced’ linear regression analyses are usually omitted in molecular test data evaluation (figure 1B). Comparison of a reference line, indicating perfect correlation between two assays (ie,  ), with a regression line enables determination of presence and magnitudes of constant and proportional error (see supplementary materials and methods) and their visualisation (figure 1B). Furthermore, the prediction intervals can be used to estimate precision of an assay in spike-and-recovery experiments (figure 1B). For SLR, one of the assays had to be chosen as the ‘gold standard’ and used as the explanatory variable, while the measurements by the second assay were treated as a response variable. Therefore, one set of measurements was assumed to have no error. This particular flaw of SLR has been previously noted, and DR was proposed as a possible solution to eliminate the issue,29 as well as other regression methods that account for presence of error in both variables.30 31
), with a regression line enables determination of presence and magnitudes of constant and proportional error (see supplementary materials and methods) and their visualisation (figure 1B). Furthermore, the prediction intervals can be used to estimate precision of an assay in spike-and-recovery experiments (figure 1B). For SLR, one of the assays had to be chosen as the ‘gold standard’ and used as the explanatory variable, while the measurements by the second assay were treated as a response variable. Therefore, one set of measurements was assumed to have no error. This particular flaw of SLR has been previously noted, and DR was proposed as a possible solution to eliminate the issue,29 as well as other regression methods that account for presence of error in both variables.30 31
Special attention should be given to the results of any combination of statistical methods used to analyse assay comparison data. BA and linear regression approaches are capable of providing similar types of information with respect to constant error, proportional error and degree of agreement estimates.32 33 However, both approaches have a number of drawbacks that should be carefully examined before using either method in isolation to make conclusions about assay performance.28 30–33 For example, the response variable directly influences the explanatory variable in BA,33 and one of the assays must be chosen as the ‘gold standard’ and used as the explanatory variable in SLR. While there is no easy solution to this problem in BA, DR can be used to eliminate the issue. This particular flaw in BA may account for the discrepancies in proportional error assessments (figures 2 and 3). Unlike SLR, DR accounts for the presence of error in both variables and is a more appropriate regression method for assay comparison studies.29 SLR, however, is a more appropriate method for spike-and-recover experiments, where a known amount of analyte can be assumed to have no error.
Conclusion
In this manuscript, BA, DR and SLR assay comparison and evaluation approaches were examined in application to quantitative aspects of NGS for molecular diagnostic assays. The utility of both BA and SLR was demonstrated by evaluating components of an NGS assay, including VAF measurements and read depth of coverage. As BA and SLR have been widely used and tested in other clinical laboratory fields, adoption of these tools by molecular laboratories can aid in evaluating, comparing and implementing emerging clinical laboratory tests such as NGS, which provide quantitative measurements for clinical tests. The use of more extensive statistical analyses would allow molecular laboratories to more fully evaluate additional performance characteristics including precision, constant error, proportional error and degree of agreement for validation and implementation of quantitative molecular assays.
Take home messages
The complementary application of Bland-Altman, Deming and simple linear regression analysis methods enables more extensive evaluation of performance quantitative characteristics of next-generation sequencing (NGS)-based assays, prior to implementation in the clinical molecular laboratory.
The application of Bland-Altman and Deming regression should be prioritised over simple linear regression for comparison studies, while simple linear regression is a more appropriate method for analysing spike and recovery data.
Bland-Altman, Deming and simple linear regression analysis methods can be used to determine additional performance metrics compared with the use of the coefficient of determination, including: constant or proportional errors, degree of agreement, precision and accuracy.
Acknowledgments
The authors would like to thank S. Garg, M. Thomas and E. Mahé for careful critiques of the manuscript.
References
Footnotes
Handling editor Runjan Chetty.
Contributors MM designed experiments, performed analyses and wrote the manuscript. MAS designed experiments, performed analysis and wrote the manuscript. VK performed analyses and reviewed the manuscript. TZ designed experiments, collected data and reviewed the manuscript. SK-R designed experiments and reviewed the manuscript. TLS designed experiments and wrote the manuscript.
Funding The Princess Margaret Cancer Foundation and Genome Canada (Genomic Applications Partnership Program).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.