Article Text

Abstract
T cell clonality testing has important clinical and research value, providing a specific and reproducible assessment of clonal diversity in T cell proliferations. Here we review the conceptual foundations of T cell clonality assays, including T cell ontogeny and T cell receptor structure and function; we also provide an introduction to T cell receptor genomics and the concept of the T cell clonotype. This is followed by a review of historical and current methods by which T cell clonality may be assayed, including current assay limitations. Some of these assay limitations have been overcome by employing next-generation sequencing (NGS)-based technologies that are becoming a mainstay of modern molecular pathology. In this vein, we provide an introduction to NGS technologies, including a review of the preanalytical, analytical and postanalytical technologies relevant to T cell clonality NGS assays.
- lymphocytes
- molecular pathology
- genetics
- oncology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Lymphocyte maturation is a fascinating process marked by immunophenotypic changes, as well as discrete and regulated molecular events.1–3 For mature T cells, an important part of this molecular ‘evolution’ involves somatic alteration of the germline configuration of the T cell receptor (TCR) genes to a unique configuration, in order to permit development of a clone of T cells with an extracellular receptor specific to a given antigen.1–3
Modern diagnostic pathology has exploited the molecular interrogation of the TCR for a number of purposes: clonality may serve as evidence that an apparently malignant lymphoid process with indefinite immunophenotypic lineage specificity is likely of T cell origin; clonality may establish a form of molecular confirmation in instances in which the ‘malignity’ of a lymphoid lesion is not certain based solely on histopathological or immunophenotypic assessment; clonality may be used as a means of minimal residual disease (MRD) testing; and clonality forms reliable proof that two seemingly distinct T cell lymphoproliferative disorders are derived from the same malignant clonal population, and to provide molecular genetic evidence of a clonal T cell infiltrate accompanying another malignant process.3–6
T cell ontogeny
T cells play a dominant role in cell-mediated immunity, recognising and eliminating a vast spectrum of antigens, and modulating the immune response by exerting control over numerous other elements of the immune system.7 T cells are postulated to originate from a common haematopoietic stem cell progenitor in the bone marrow, whereafter they home to the thymus.8 It is in the thymus that T cells pursue a series of maturation steps by which antigen specificity is established, as well as exclusion of self-specific antigenicity.7 T cell antigen specificity is mediated through the TCR. The thymus contains epithelial and antigen-presenting cells responsible for exposing naïve T cells to a variety of antigen presented in the cleft of the major histocompatibility complex molecule (MHC).7 TCRs with absent avidity to self-antigen-MHC molecules (ie, unable to recognise any antigen presented in MHC), as well as TCRs with strong avidity to self-antigen-MHC molecules (ie, likely to cause an immunological response to one’s own tissues), will die within the thymus.7 Once the T cell maturation process is complete, the now-mature but non-self-antigen naïve T cells will home to lymphoid tissues to await subsequent antigenic stimulation.7
Basic structure of the TCR
The TCR is a heteroduplex molecule anchored to the external aspect of the cell surface where, in cooperation with numerous additional signalling and structural proteins, the TCR functions to recognise an antigen with a high degree of specificity.9 10 This specificity, and indeed the vast array of potential antigenic epitopes recognisable by the population of T cells on the whole, is afforded by (1) the number of TCR encoding regions of a given TCR’s genes as present in the germline; and (2) the intrinsic capacity of the TCR gene loci to undergo somatic rearrangement.3
Unlike immunoglobulin receptors, able to recognise an antigen in its natural form, TCRs typically recognise antigen within the context of an MHC molecule.11 Thus, specific regions of the TCR capable of recognising MHC are conserved, defined by the complementary determining regions 1 and 2 (CDR1 and CDR2, respectively); the region of antigen specificity is determined by the CDR3 region.11 Of note, the CDR3 is invariably recognisable by flanking amino acid motifs, namely the downstream F/W-G-X-G amino acid motif and the upstream second cysteine.12 These invariant sequence motifs contribute to the formation of the so-called ‘Collier-de-Perles’, the higher order structure required for antigen specificity.12
There are four types of TCR protein monomers in humans, the α, β, γ and δ proteins. These combine to form functional TCRs composed of α/β or γ/δ subunits.9 13 The vast majority (>95%) of circulating T cells are of the α/β type,9 13 and whereas most T cells of the α/β subtype home to lymphoid-predominant tissues (ie, lymph nodes and spleen), the γ/δ subtypes have a propensity for epithelial tissues.13 This seeming anatomical specificity of γ/δ T cells is hypothesised to relate to a more ‘innate immunity’ function of γ/δ T cells relative to α/β T cells.13 The α proteins are encoded by genes in the T cell receptor alpha (TRA) locus; the β proteins by genes in the T cell receptor beta (TRB) locus; the γ proteins by genes in the T cell receptor gamma (TRG) locus; and the δ proteins by genes in the T cell receptor delta (TRD) locus.
Genomic organisation and germline configuration of the TCR genes
The human TCR genes are organised into four loci: TRA, TRB, TRD and TRG. The TRA locus is found on the long arm of chromosome 14 in band 14q11.2 and spans a total of 1 megabase.14 Sandwiched between the TRA V and J domains is the TRD locus (14q11.2), itself spanning only 60 kilobases (kb).15 The TRB locus is found on the long arm of chromosome 7 in band 7q35 and spans a total of 620 kb.16 The TRG locus is found on the short arm of chromosome 7 in region 7p15-p14 and spans 160 kb.17 Interestingly, the organisation of TCR genes into four TCR loci seems invariably common to all jawed vertebrates, despite distinct specific genomic loci.18 Indeed, all tetrapods studied thus far have a comparable organisation of the TRD locus within the TRA locus, but with separate loci for the TRB and TRG genes.18 Many of the genes themselves have a surprising degree of sequence-level conservation between species.18
Within each TCR gene locus are a number of variable (V) and join (J) segments14–17; additional diversity (D) segments are present within the TRB and TRD loci.15 16 These V, D and J segments are grouped into respective V, D and J regions (figure 1). In the germline configuration, a full complement of V genes (numbering from as few as 8 in TRD to as many as 54 in TRA), D genes (2 in TRB and 3 in TRD) and J genes (numbering as few as 4 in TRD to as many as 61 in TRA) can be detected, varying in number based on inheritance.14–17 The specific TCR sequences themselves may also vary person-to-person based on the alleles inherited; these are variable in number for a given gene and represent sequence polymorphisms.14–17 The TCR germline configuration gene sequences and their associated alleles are documented in detail in the ImMunoGeneTics (IMGT) database.12 This database has been the primary curator of TCR, immunoglobulin and MHC sequence data for nearly three decades, working in partnership with leading global molecular biology organisations such as the European Molecular Biology Laboratory.12
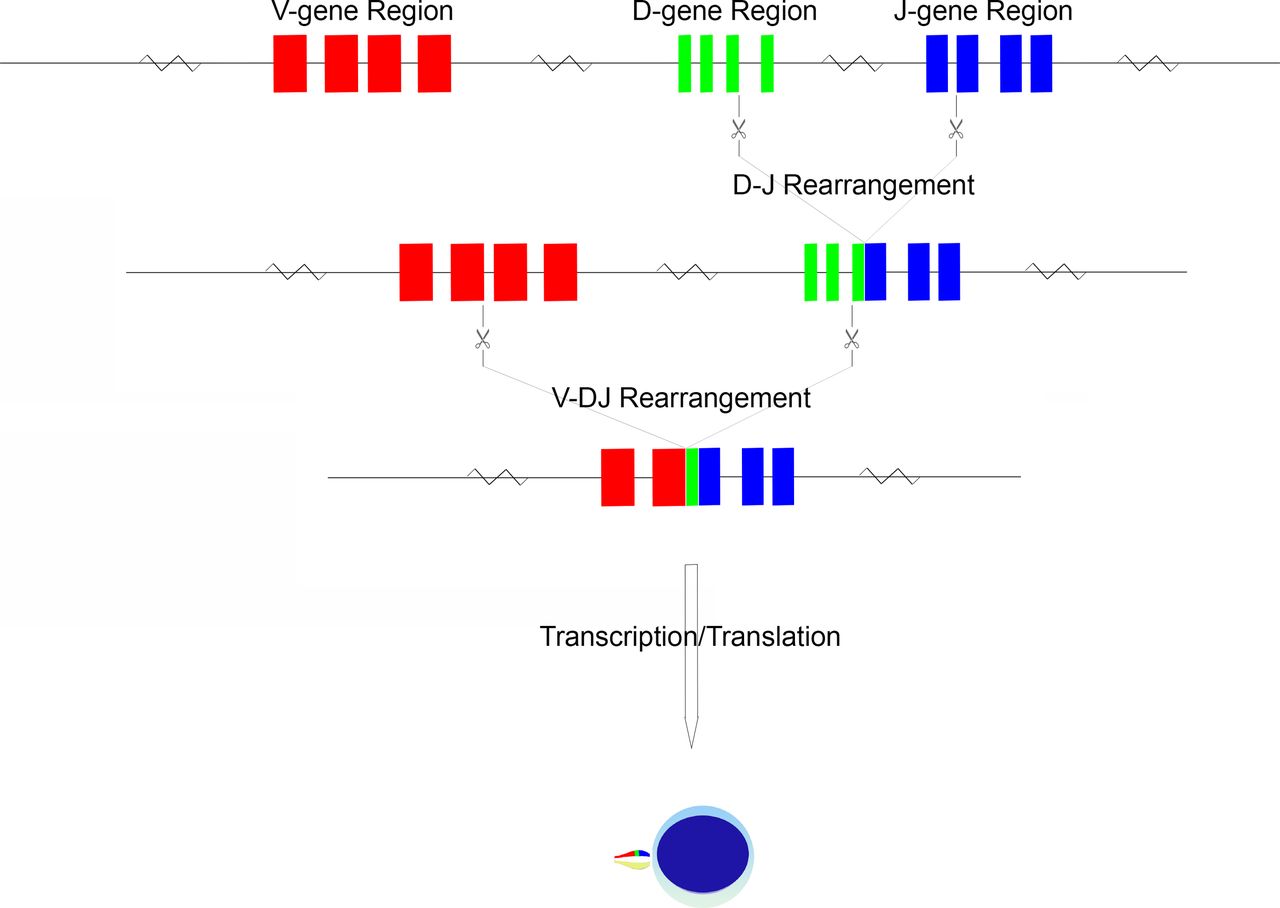
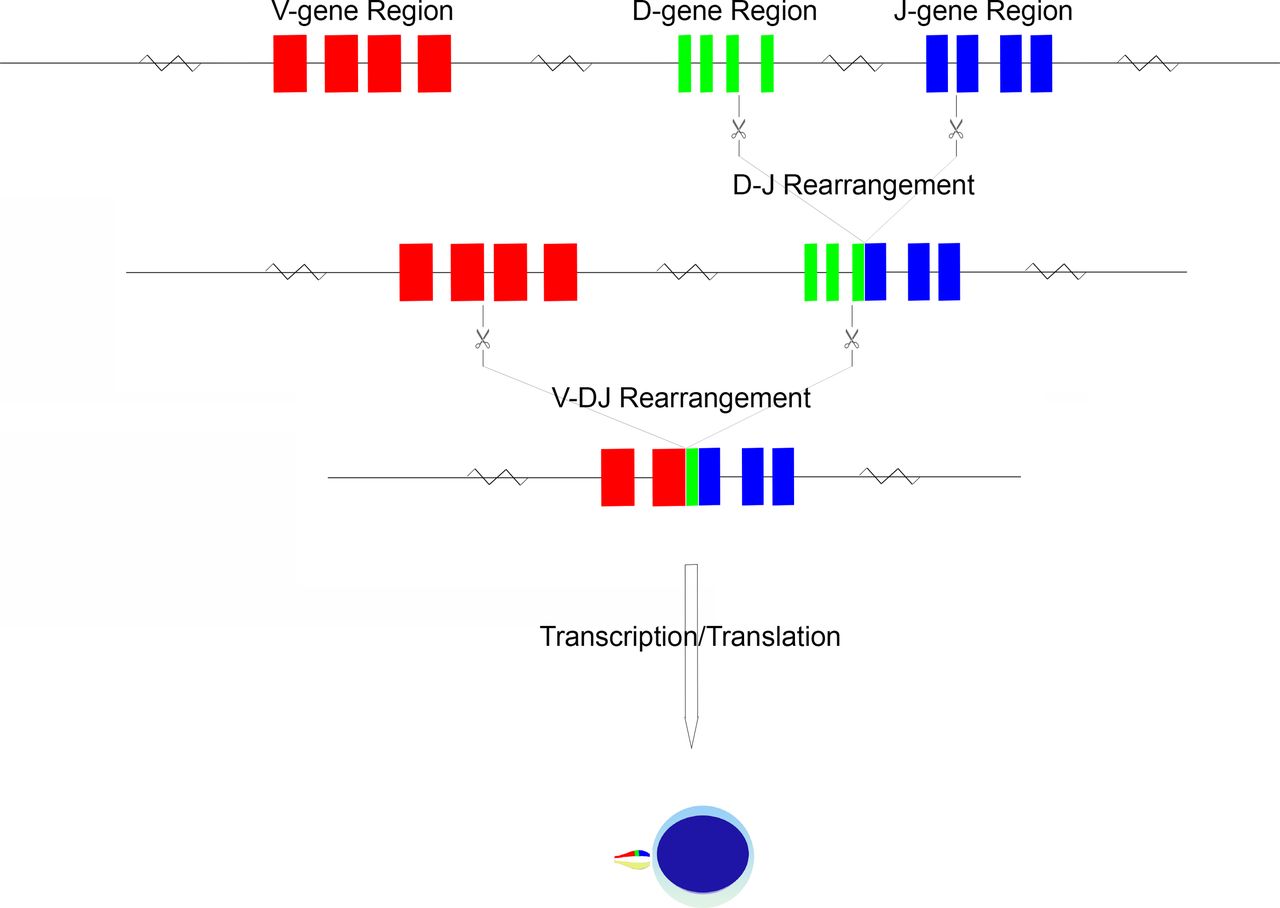{kind=link}
Schematic of TCR gene rearrangement. The schema begins in the germline configuration, followed by D-J gene rearrangement and finally by V-DJ gene rearrangement. The rearranged TCR configuration is then transcribed and translated into a protein monomer, which is paired with its monomer protein counterpart to form a functional TCR.
During T cell maturation, there is recombination of genes within the V, D and J regions to produce a lineage of cells with a novel configuration of TCR genes.7 This gene rearrangement, after gene transcription and translation, produces a TCR unique to a given T lymphocyte lineage. Rearrangement is driven by series of DNA splicing reactions directed to sites flanking the genes of interest; the sites of cleavage are indicated by recombination signal sequences (RSS), consisting of conserved heptamer and nonamer sequences.19 By recognition of the flanking RSS, the recombination machinery forms hairpin loops of undesired intervening sequence, thus approximating V-D and D-J genes as required for the specific locus.19 This splicing is imprecise, with frequent introduction of small deletions or insertions at the sites of cleavage.19 Non-templated nucleotides may be inserted at junction sites (so-called n-nucleotides), a process mediated by terminal deoxynucleotide transferase.20 Non-random insertion may also occur owing to a nick-hairpin-ligation process whereby short palindromic segments are produced (so-called p-nucleotides).21 It is this inexactness that has the desirable effect of introducing sequence variation at the V-(D)-J junction sites, and hence the potential for amino acid variation at the site of antigen recognition, namely the CDR3 region.
An ordered progression of rearrangement has been identified, beginning with TRD, followed by TRG, TRB and finally TRA.22 Once a rearrangement has occurred, allelic exclusion prevents further rearrangement of the locus as well as that of the other chromosome23; this process of allelic exclusion appears to rely on feedback from β chain production, perhaps mediated by the p56lck non-receptor protein tyrosine kinase, resulting in downregulation of the gene rearrangement machinery.23 Thus, a T cell population evaluated at the genomic level may have several identifiable combinations of TRA, TRB and TRG gene rearrangements. Indeed, α/β type T cells will have identifiable (but non-expressed) TRG gene rearrangements. These additional non-functional or non-expressed rearrangements can serve as additional parameters in the definition of a T cell populations TCR genomic fingerprint.
The concept of the TCR clonotype
TCRs are not subject to somatic hypermutation when a given T cell is exposed to antigen.11 As such, the specificity of a given T cell clone remains static once rearrangement has occurred; this common antigen specificity is referred to as the clonotype of that population.24 A T cell population’s clonotype is a molecular description of the unique sequences (typically described from the amino acid perspective) required to produce the T cell’s TCR antigen specificity, as well as the specific V and J genes involved in the composite rearrangements. It should be noted that the D genes, where applicable, are subsumed within the CDR3 sequence region in order to produce the characteristic antigen specificity; this forgoes the need to include a named D-gene in the clonotype description. Thus, the CDR3 regions can be regarded as k-mers (subsequences of either nucleotides or amino acids, of length k) of the full TCR component subunits.
Given that stepwise rearrangement of all four TCR loci is possible in a given T cell clone, definitive TCR clonotyping requires sequencing of rearranged TCR gene transcripts.24 TCR clonotyping using genomic DNA, however, is much more frequently performed5; in such instances, the definitive TCR clonotype is subsumed in a broader library of possible clonotypes. Such an approach is often simpler and is not subject to the potential confounder of expression levels. Furthermore, when combined with cell-sorting technologies, a definitive TCR clonotype might be inferred from an adequately uniform population of T cells.5
Development of T cell clonality testing
Routine TCR clonality testing began in the 1980s,1 at which time several publications noted relevance to the diagnostic work-up of T cell lymphoproliferative disorders.25 26 Early work also highlighted the specificity of TCR clonality testing for T cell malignancies in general, when the context was clearly a lymphoid malignancy, but of uncertain lineage.1 More recently, the use of T cell clonality for detection of MRD has been touted as a highly sensitive and specific tool,27 for example in cases of suspect residual/recurrent T cell malignancies post-treatment. Other evidence has shown that T cell clonality assessment is a useful adjunct to the work-up of locoregional lymph node involvement by cutaneous T cell lymphoma, both as a diagnostic and prognostic modality.28
The first TCR clonality assays employed the use of restriction enzyme digestion of query DNA, followed by gel electrophoresis and Southern blotting using probes for the known TCR genes.29 Although cumbersome, this approach is still considered by some to be the gold-standard T cell clonality assay.29 More recently, PCR-based assays have become the workhorse of clonality assessment; these are advantageous in that they do not require as much input nor necessitate high-quality DNA, are faster than Southern blot-based assays and can provide very sensitive quantitative measures of clonal rearrangements; the latter has permitted the use of clonality testing (both immunoglobulin and TCR) for the purposes of MRD assessment.29 More recently, internationally accepted PCR primer sets have been introduced to further standardise PCR-based T cell clonality assays.
The primers most frequently used in PCR-based TCR clonality analysis, termed the BIOMED primer set,6 are the product of several years of collaborative expert study, resulting in a thoroughly studied consensus T cell clonality assay primer set. Commercially available BIOMED assays include multiplexed primer sets for both immunoglobulin and TCR clonality assessment and can be implemented using equipment widely available in clinical laboratories (eg, fluorescent capillary electrophoresis platforms).6 These commercially available primer sets, typically multiplexed as two to three reactions per locus, have the advantage of standardisation and ease of implementation. In addition, by virtue of the extensive study performed by the BIOMED consortium (now known as the EuroClonality division of the European Scientific foundation for Laboratory HematoOncology), the BIOMED assay has the well-documented advantage of capturing the rearrangement patterns of the vast majority of study cohort lymphomas bearing productive TCRs.6 Positive (monoclonal) and negative (polyclonal) controls are included with each assay run; these permit the confirmation that the PCR reactions have occurred and provide useful reference electropherographic patterns to which query samples may be compared.
Limitations of current T cell clonality assays
The current BIOMED approach to TCR clonality testing is subject to a number of technical and practical caveats.30 Because the PCR-based techniques that are employed in T cell clonality assays are subject to amplicon size restrictions,6 31 the sheer size of the TRA locus prevents a complete assay of the TRA gene in clinical settings. Indeed, although of smaller size, the TRB locus as a whole is also quite difficult to assess in its germline configuration, even when a multiplexed PCR strategy is employed. Thus, only a subset of the TRB locus is covered in the BIOMED multiplexing strategy.6 It is therefore of no surprise that much of the published data pertaining to the utility and validity of T cell clonality assays have stemmed from the only subparts of TRB as well as the much smaller and more condensed TRG locus. In addition, since the TRD locus is often deleted after TCR gene rearrangement (as it is contained within the TRA locus and excised whenever the TRA locus is rearranged), assays for TRD have also not been as rigorously studied. For this reason, any BIOMED-based T cell clonality assay aimed at fully describing a TCR clonotype will be insufficient.
The BIOMED assay is subject to additional interpretative challenges. As part of the standard T cell clonality assay, most laboratories rely on the demonstration of electrophoretic migration patterns for the determination of clonality: a prototypical ‘polyclonal’ T cell population will produce an electropherogram with a Gaussian distribution of low-fraction amplicon peaks; in contrast, a bona fide ‘monoclonal’ T cell population will produce an electropherogram with a dominant high-fraction single amplicon peak. Thus, interpretation of the assay depends on the demonstration (or lack thereof) of a dominant amplicon of specific (although not necessarily predefined) molecular weight, rather than a Gaussian distribution of a mixture of amplicons. This approach, as has been described previously,32–34 may be subject to interpretative error; a minor clone, for example, may be masked by a noisy background population.
Finally, current clinical TCR clonality assays often require relatively large amounts of input DNA. This relates to the need for a recommended input of 50–100 ng of DNA to multiple individual reactions.6 Specifically, when the TRB locus is being assessed, the multiplexed PCR approach requires three separate primer combinations; in concert, a TRG assessment will require two separate primer set experiments.6 Thus a typical clinical assessment of five separate primer sets will require a minimum of 250–500 ng of input DNA.
Background to next-generation sequencing (NGS) technologies
Massively parallel DNA sequencing technologies, often termed ‘next-generation sequencing’, have enabled measurement of millions of segments of the genome simultaneously and permit parallelisation of DNA sequencing with much higher throughput than traditional sequencing technologies.35 Naturally, these technologies are also ideally suited for a movement away from assays based on fragment size and towards sequence-level resolution of TCR rearrangements. To facilitate the effective use of these technologies, an understanding of the preanalytical (ie, DNA library preparation and signal enrichment), analytical (ie, sequencing instruments) and postanalytical (ie, bioinformatic analysis) considerations is required.35 36
Library preparation
Library preparation involves the processing of input DNA into a form evaluable by an NGS instrument. This requires that DNA be isolated and purified, typically followed by a shearing step in which enzymatic, chemical or mechanical forces are applied to fragment the DNA into smaller sizes.36 While this process produces smaller, more manageable-sized fragments, it also tends to damage the ends of the DNA, for which an end-repair process is required.36 Subsequently, adapter ligation is performed; adapters consist of oligonucleotides of prespecified sequence that are sequencing platform-specific and used to assist in sample multiplexing, priming for amplification, or to facilitate sequencing-by-synthesis (SBS) and DNA strand immobilisation to sequencer-specific media.36 A wide variety of commercial library preparation kits are available for many of the different sequencing platforms, including integration of unique molecular identifiers and tagging of large, contiguous stretches of the genome.
Target enrichment
Target enrichment involves the selective amplification of sequencing signals from the portion of the genome of interest, in excess of the remainder. The two most frequently employed techniques are amplicon-based target enrichment, by which PCR is directed (usually in a multiplexed manner) to regions of interest; and hybrid capture-based target enrichment, by which probes complementary to regions of interest are used to physically capture the sequences of interest.37 Amplicon-based and capture-based techniques each has previously studied pros and cons: amplicon-based approaches are fairly easy to perform, incorporate familiar and automatable technologies, but are often restricted to relatively small overall target sizes when multiplexing is desired.37 In contrast, while capture-based methods require a slightly more complex workflow, they are able to scale up for enrichment of larger fractions of the genome.37 A variety of manufacturers of hybrid capture kits exist, some exploiting RNA–DNA hybridisation (eg, Agilent SureSelect kits), while others employ DNA–DNA hybridisation (eg, IDT xGen probes). Furthermore, some hybrid capture systems incorporate solid-phase capture media (akin, eg, to a microarray), whereas (more commonly in the present) capture is performed in solution using biotinylated hybridisation probes, which can then be precipitated from solution using paramagnetic streptavidin-coated beads.37
Sequencing platforms
A variety of NGS platforms exist, most employing an SBS approach.36 The primary sources of interplatform variation relate to the sequencing reaction media, chemistry and the methods of detection.36 Illumina platforms, for example, exploit reversible dye-terminator sequencing within a special flow cell: input DNA is injected into the Illumina flow cell wherein it anneals to a ‘lawn’ of library adapter-specific oligonucleotides; a series of PCR amplification steps then produces clusters of cloned short-sequence DNA; during the sequencing cycle, individual bases with characteristic fluorescence are then enzymatically added with each cycle of strand extension; the resulting fluorescence patterns are used to decipher the oligonucleotide sequence within each flow cell cluster.36 In contrast, Ion (Thermo Fisher) platforms exploit pH changes (which are specific to the given base added during SBS) occurring during the polymerisation reaction for detection.36 In addition, the Ion platforms employ emulsion PCR rather than the solid-phase approach used in the Illumina flow cell.36 The PacBio platform (Pacific Biosciences) employs nanophotonic reaction chambers, each containing a fixed polymerase enzyme. SBS is then performed, also employing fluorescence-tagged nucleotides. In contrast to the Illumina and Ion platforms, which perform massively parallelised, short-read sequencing, the PacBio platform technology permits sequencing of oligonucleotides of up to several kilobases in length. Still other platforms exist in which sequencing by direct sequence interrogation is performed, rather than SBS; the Oxford Nanopore platform, for example, functions by passing long single-stranded DNA molecules through carefully constructed protein nanopores, during which voltage changes are registered and translated to specific nucleotide sequences.38 It should be noted, however, that many of these technologies are manufacturer-branded, research use-only; indeed, only a small subset of Illumina and Ion platforms are Food and Drug Administration-approved for clinical use.
Commercially available NGS-based T cell clonality assays
A number of commercially available NGS-based T cell clonality assays exist, with three companies appearing to represent the largest commercial NGS T cell clonality assay market share. Adaptive Biotechnologies is a recently founded US-based company offering both kits and reference-lab testing of both TCR and immunoglobulin genes. Adaptive offers both research (ImmunoSeq) and clinical-grade (clonoSEQ) amplicon-based assays to interrogate the TCR and immunoglobulin genes. Adaptive’s assays are advertised as being sufficiently sensitive for the purposes of MRD assessment, as well as sufficiently multiplexed to permit analysis of all four loci. Adaptive has addressed the issue of informatics by way of proprietary analysis pipelines and visualisation tools. Invivoscribe has a lengthier history of TCR and immunoglobulin clonality assay development, having provided BIOMED-based kits for several decades. Recent assay redevelopment has permitted compatibility with both MiSeq and Ion platforms, by incorporating Illumina and Ion compatible adapters, respectively. 10x Genomics focuses mainly on single-cell/single-DNA molecule sequencing technologies. Using micellisation, input cells can be independently indexed and subjected to library preparation. Individual micelles are then pooled and sequencing proceeds as usual. The theoretical advantage to the 10x Genomics approach is the potential to identify the functional TCR clonotype of a given cell, without inference from a large pool of detected rearrangements.
Laboratory-developed tests and novel approaches to T cell clonality testing
Laboratory-developed tests (LDTs) have played an important historical role in T cell clonality testing, especially in the early decades of testing using restriction-fragment polymorphism analysis and ‘homebrew’ primer sets. While the recent decades of standardisation have reduced the LDT footprint, especially by virtue of the work of the BIOMED consortium and the increasing availability of commercial kits, a number of laboratories continue to champion ‘in-house’ test design as more affordable and flexible. One relatively user-friendly LDT design approach, currently being tested in our laboratories in Toronto, Ontario, and Calgary, Alberta, is TCR and immunoglobulin gene clonality testing using a hybrid capture approach to target enrichment. Central to these efforts is the hypothesis that a hybrid capture enrichment protocol, with probes designed against the germline TCR gene sequences, might sufficiently enrich rearranged TCR loci. By aligning the subsequent NGS data to the germline TCR genes, reads containing subsequences aligning to both V and J genes could be taken as evidence of a rearrangement. By taking a tally of such reads over the entire data set, and recording the named TCR genes as well as regular expression identification of CDR3 sequences, a full picture of the TCR clonotype could be generated. Early data (pending publication) suggest comparable sensitivity and specificity relative to current standard clinical assays.
Bioinformatic considerations
Although many of the commercial TCR clonality assay kits include informatics support, those laboratories choosing an LDT approach to NGS-based T cell clonality testing will require either third-party informatics support or (perhaps more likely) will need to develop their own in-house pipelines. Fortunately, there already exist some freeware solutions to assist in the development of in-house solutions. The IMGT provides a freely accessible TCR/immunoglobulin gene rearrangement analysis tool for use with NGS data (IMGT/HighV-QUEST),39 which provides users with read-for-read clonotype assessment (including named V, D and J genes and alleles, as well as the CDR3 region DNA and amino acid sequences). The recently published MiXCR pipeline provides similar data to that of the IMGT/HighV-QUEST, with the added advantage of filtering algorithms.40 These algorithms define the so-called ‘core-clonotypes’, which effectively represent dominant gene and CDR3 sequence combinations, with low-frequency clonotypes corrected to the most likely core clonotype pattern.4 These bioinformatic postsequencing correction algorithms may be useful to users working with relatively homogeneous populations, but could theoretically result in data loss in highly heterogeneous T cell populations or in the context of MRD assessment. Finally, our own laboratories are working to develop a TCR/immunoglobulin clonality assay data analysis pipeline with the additional feature of a clonotype-specific predictive cell fraction algorithm.
Conclusion
T cell clonality assays have a lengthy history of utility in both clinical and research settings. TCR assays provide molecular genetic evidence of clonality in malignant or suspect lymphoproliferative disorders and can facilitate MRD assessment.3–6 Recent work has led to improvements in T cell clonality assays with the application of NGS technologies, and progress continues. Beyond the value of T cell assessment in the context of T cell neoplasia, however, T cell clonality assays are of increasing interest in the field of T cell immunotherapy.41 In this context, the identification of a tumour-specific T cell clone, typified by a specific T cell clonotype, can be critical to the success and monitoring of therapy.41 Given the need for sequence-specific data to accurately define a T cell clonotype, and the potential heterogeneous T cell population that might exist in association with a tumour, it follows logically that a high-throughput and multiplexed approach to T cell clonotyping would be most useful, likely afforded via application of NGS-based technologies. It is conceivable that future clinical need for T cell clonality assessment will be dominated by immunotherapeutic indications, rather than diagnostic ones.
Take home messages
TCR assays provide molecular genetic evidence of clonality in malignant or suspect lymphoproliferative disorders and can facilitate MRD assessment.
TCR assay development and validation has a lengthy history, resulting in consensus primer sets that form the standard in most countries.
The need for sensitive and sequence-specific data, especially to direct and monitor T-cell immunotherapy, portends the utility of high-throughput next-generation based sequencing assays.
Acknowledgments
We would like to recognise the support and assistance of the members of the Princess Margaret Advanced Molecular Diagnostics Laboratory, University Health Network Pathology Genome Diagnostics Laboratory and the Princess Margaret Genomics Centre (www.pmgenomics.ca).
References
Footnotes
Handling editor Runjan Chetty.
Contributors All authors have contributed to, reviewed and approved of the manuscript.
Funding EM was supported by a Terry Fox Foundation/Canadian Institutes of Health Research Fellowship and is currently supported by Calgary Lab Services and the University of Calgary Hematology Research Grants. TP’s work related to immune cell clonality testing is supported by grants from the Princess Margaret Cancer Foundation, Canada Foundation for Innovation, Canadian Cancer Society Research Institute, Terry Fox Research Institute, the Multiple Myeloma Research Foundation, and the Ontario Institute for Cancer Research. SK-R has received funding from Pfizer Canada and from the Princess Margaret Cancer Foundation.
Competing interests TP and EM are listed as inventors on a patent for profiling immune cell repertoires using hybrid capture and next-generation sequencing methods. SK-R has not competing interest to declare.
Provenance and peer review Not commissioned; externally peer reviewed.