Article Text

Abstract
Aims Peripheral T cell lymphoma not otherwise specified (PTCL/NOS) is the commonest PTCL subtype. Recently, proliferation pathways have been found to be commonly altered in PTCL/NOS. CDKN1B/p27, a critical regulator of cell cycle and proliferation, has been suggested to be involved in T cell lymphomagenesis. This study aimed to evaluate the possible occurrence of CDKN1B/p27 aberrations in PTCL/NOS.
Methods CDKN1B/p27 expression at RNA and protein level by DNA and tissue microarrays, in 28 and 98 cases, respectively, was studied. Additionally, direct sequencing of CDKN1B in 81 PTCL/NOS was performed.
Results CDKN1B mRNA was similarly expressed in PTCL/NOS and normal T lymphocytes. In addition, structural abnormalities were not found; these included mutations and deletions in any exons, exon–intron junctions or regulatory regions. Furthermore, physiological expression of p27 in neoplastic cells was demonstrated by immunohistochemistry; this was mutually exclusive with Ki-67, as expected when the system is intact. Consistently, the expression of other molecules that are functionally related to CDKN1B/p27 in controlling cell cycle (including CCNE1) did not appear to be affected at either the mRNA or protein level. Finally, it was found that p27 expression was not significantly related with overall survival.
Conclusion CDKN1B/p27 aberrations seem to be uncommon in PTCL/NOS pathogenesis.
- CDKN1B/p27
- gene sequencing
- haematopathology
- immunohistochemistry
- lymphoma
- microarray
- molecular genetics
- peripheral T cell lymphoma
Statistics from Altmetric.com
- CDKN1B/p27
- gene sequencing
- haematopathology
- immunohistochemistry
- lymphoma
- microarray
- molecular genetics
- peripheral T cell lymphoma
Introduction
Peripheral T cell lymphomas (PTCLs) correspond to a heterogeneous group of tumours basically subdivided into specified and not otherwise specified (NOS).1 While specified tumours are distinct but rare entities, PTCL/NOS represents the commonest subtype, being characterised by variable morphology, usually aberrant immunophenotype, and aggressive clinical course.1 Notably, the molecular pathology of this tumour is poorly understood.2 In fact, many alterations have been found, but no single genes have been demonstrated to have a pathogenetic role.2 Recently, gene expression profiling (GEP) studies have led to the identification of PTCL/NOS-associated signatures, allowing a better definition of their histogenesis, pathogenesis and prognostication.3–5 In particular, proliferation has been found to be commonly altered in PTCLs,5 6 with the proliferation rate being a well-defined adverse prognostic factor.6 7
Cyclin-dependent kinase inhibitor 1B (p27), encoded by CDKN1B gene, is a member of the cyclin-dependent kinase inhibitors.8 Cyclin-dependent kinase inhibitors inhibit cyclin-dependent kinases (CDKs) or CDK–cyclin complex activities, thereby resulting in proliferation arrest. Specifically, p27 physiologically controls the cell cycle progression by limiting G1–S transition8 through its activity as a potent inhibitor of cyclin E and cyclin A complexed with CDK2.9
In resting lymphocytes, p27 is usually expressed, while it is downmodulated in activated lymphocytes. Consistently, in normal lymphoid tissues, p27 and the proliferative marker Ki-67 are mutually exclusive.10
Importantly, CDKN1B/p27 alterations actually result in loss of cell cycle control and possible malignant transformation. In fact, variations of p27 expression have been associated with several tumours,11 including different non-Hodgkin lymphomas (NHLs).12–14 Generally, in such instances, CDKN1B/p27 abnormalities have been referred to specific single-nucleotide polymorphisms (SNPs), point mutations, epigenetic events, and loss of function due to cyclin-E1 (CCNE1) overexpression.15–18 Noteworthy, higher p27 levels have been found in a proportion of resting neoplastic cells refractory to chemotherapy and radiation, while lower levels of p27 characterise the proliferative fraction.8 15 19 Thus, patterns of expression and functional role of p27 in cancer development have been found to be complex and are not completely understood. As far as NHLs are concerned, indolent lymphomas such as follicular and chronic lymphocytic leukaemia, as well as mantle cell lymphoma, are often associated with p27 overexpression,14 20 while other aggressive lymphomas generally present with lower protein levels.21–23 These findings suggest that low p27 expression might be a prerequisite for sustained high proliferation; this concept is supported by different studies.24
Interestingly, it was shown recently that p27 deficiency can cooperate with BCL2 overexpression in promoting T cell precursor lymphomagenesis in mice,25 indicating that p27 is a critical tumour suppressor in the context of Bcl-2 production. However, data on PTCLs have so far been limited to small series, mainly consisting of anaplastic large cell lymphomas (ALCLs),16 21 26 and specific information on PTCL/NOS is not available.
The present study was designed in order to identify possible determinants of abnormal proliferation in PTCL/NOS by specifically focusing on the expression and molecular structure of p27 in a large series of PTCLs/NOS.
Materials and methods
Gene expression analysis
We analysed GEP data of 28 PTCL/NOS derived from cryopreserved lymph nodes, 4 non-neoplastic reactive lymph nodes, and 20 samples of normal T lymphocytes (CD4+, n=5; CD8+, n=5; HLA-DR+, n=5; HLA-DR–, n=5), previously generated by using the Affymetrix HG-U133 2.0 plus microarray (Affymetrix, Santa Clara, California, USA) and available at the National Center for Biotechnology information (http://www.ncbi.nlm.nih.gov/projects/geo/). For technical details, see reference.5 In particular, we focused on the expression of CDKN1B and other molecules, such as CCNE1, CCND1, CDk2, CDk4, CDk6 and TP53, that are functionally related to CDKN1B in controlling the cell cycle.
CDKN1B normalised values were compared in PTCLs versus normal samples by using a Student t test.
Immunohistochemistry on tissue microarrays
Furthermore, we studied the expression of p27 and CCNE1 by immunohistochemistry (IHC) on tissue microarrays (TMAs) containing 98 PTCL/NOS cases, collected from lymph nodes of patients who had been diagnosed according to the WHO classification.1 In particular, p27 expression was tested by a mouse monoclonal antibody, clone F-8, (Santa Cruz Biotechnology, Santa Cruz, California, USA) dilution 1:10, while CCNE1 expression was tested by a mouse monoclonal antibody, cyclin-E Ab-5 (clone 13A3) dilution 1:40 (Thermo Scientific, Waltham, Massachusetts, USA). A rabbit anti-Ki-67 monoclonal antibody (clone SP6, dilution 1:100; Thermo Scientific) was used to evaluate the proliferative index. Ki-67 staining was scored as previously reported,6 by identifying three subgroups (<20%, 20–80% and >80% stained cells) The sections underwent antigen retrieval in 1 mM EDTA buffer (pH 8.0) in a microwave oven at 900 W (three cycles lasting 5 min each) and revealed by the EnVision and APAAP (Dako, Golstrup, Denmark) techniques.6 Cores were considered positive if 30% or more of the tumour cells were stained.6 Ten reactive non-neoplastic lymph nodes were used as controls.
In addition double staining was performed on 6/98 PTCL/NOS to test the possible p27 and Ki-67 co-expression in the same population. The above-mentioned antibodies were used in association with two secondary antibodies: Alexa Fluor G488 goat anti-mouse IgG1 (p27) and Alexa Fluor R568 goat anti-rabbit IgG (Ki-67) (Invitrogen, Eugene, Oregon, USA).
Gene sequencing
Genotypes were determined by using PCR amplification and direct sequencing. In particular, PCR products of all the three exons, the exon–intron junctions and the regulatory regions of CDKN1B were directly sequenced in 81/98 PTCL/NOS also used for the TMAs. Primers and relative conditions of amplification were kindly provided by Dr Bao-Li-Chang and Dr Jianfeng Xu (Center for Human Genomics, Wake Forest University School of Medicine, Wiston-Salem, North Carolina, USA) (for details, see table 1, in Supplementary File 1).17
Statistical analysis
Statistical analyses were carried on with the StatView 5.0 software package (SAS Institute Inc, Cary, North Carolina, USA). Student t test was adopted for GEP data analyses and for p27 expression evaluation according to the presence of T326G SNP. Survival data were analysed by using the Kaplan–Meier method and Cox regression. The limit of significance for all analyses was defined as p<0.05; two-sided tests were used in all calculations.
All cases were collected at diagnosis, before any treatment administration. Clinical information and complete follow-up were available in 67/98 cases. All patients gave permission for use of samples for genomic research and the study was approved by the local ethics committee.
Results
Gene expression analysis
First, we investigated whether CDKN1B levels were different in different T cell subsets. Indeed, as expected, we found that resting T cells presented with higher CDKN1B expression in comparison with activated ones. However, the difference was not significant (p=0.3; not shown), probably due to the high degree of variability among samples. Accordingly, we compared the tumour cases with the entire T cell population (activated, resting, CD4+ and CD8+). Interestingly, GEP showed no significant differences in CDKN1B expression when PTCL/NOS cases were compared with non-neoplastic reactive lymph nodes and normal T lymphocytes (figure 1). In particular, the median expression values were similar in the two groups, with only scattered normal samples showing levels higher than the maximum value recorded in the tumour samples. Importantly, in order to avoid the effects of possible reactive components admixed to neoplastic cells, which may give confusing results, we normalised the expression values to the percentage of tumour cells, as evaluated by two expert haematopathologists using morphology and immunohistochemistry.

CDKN1B expression in peripheral T cell lymphoma not otherwise specified (PTCL/NOS). Expression values of CDKN1B in cases of PTCL/NOS versus samples of normal tissues evaluated by gene expression analysis on DNA microarrays. Median values are indicated by red lines. No significant differences in CDKN1B expression were observed in PTCL/NOS cases compared with normal tissues (2212.87 vs 2215.5, p=0.99).
In addition, we investigated the expression of genes that are functionally related to CDKN1B/p27 in controlling the cell cycle (ie, CCNE1, CCND1, CDk2, CDk4, CDk6 and TP53), and did not find any consistent abnormality (not shown).
Immunohistochemical analysis
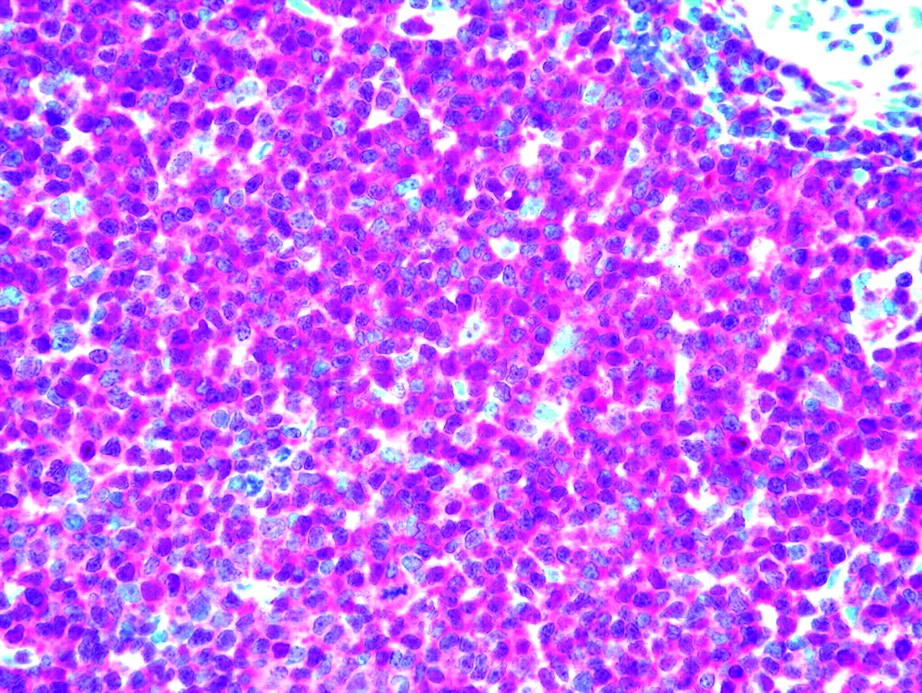
Second, we studied the immunohistochemical expression of p27, CCNE1, and Ki-76 in 98 PTCLs/NOS on TMAs. In particular, p27 was expressed in 46/98 cases (47%) (figure 2). On the other hand, Ki-67 expression was ≥80% in 6/98 (6%), ≥20% and ≤80% in 74/98 (76%), and ≤20% in 18/98 (18%) cases. Notably, the six PTCLs/NOS with Ki-67 ≥80% presented variable degrees of p27 expression (range 10–35%). Thus, we further investigated these cases by double-staining immunofluorescence in order to establish whether p27 and Ki-67 could be co-expressed by the same cells. Interestingly, the two molecules were not coexpressed (figure 3) as expected for p27 physiological activity. Furthermore, we studied CCNE1 expression, as this molecule has been reported to possibly overcome p27 function in some cancers.8 15 However, we found no CCNE1 expression in PTCLs/NOS.
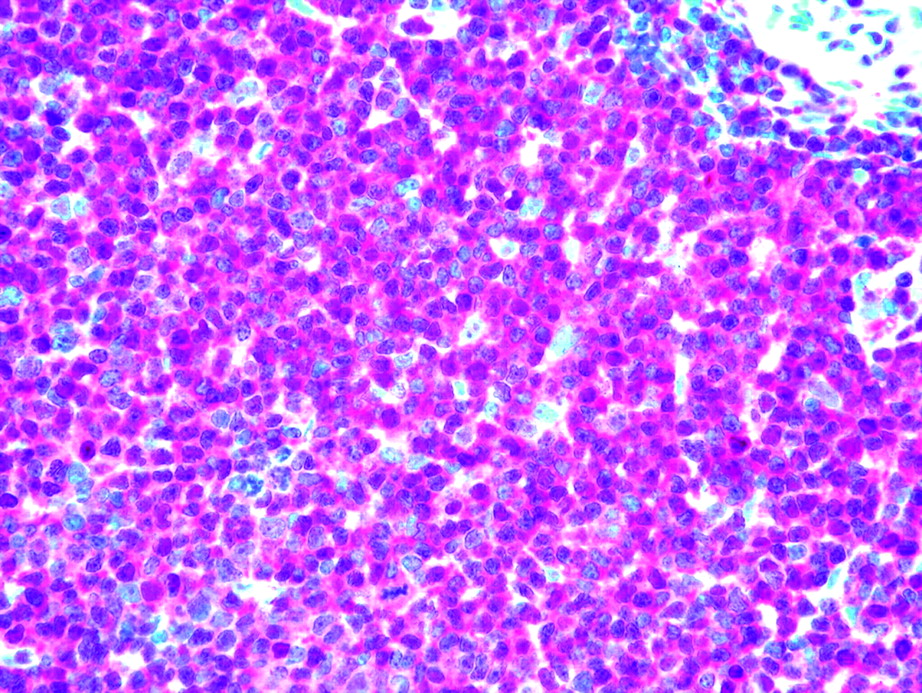
CDKN1B/p27 expression in peripheral T cell lymphoma not otherwise specified (PTCL/NOS). CDKN1B/p27 immunostaining in a PTCL/NOS sample (APAAP technique; Olympus BX41 microscope, Olympus CAMEDIA C-7070 camera, magnification ×400; colours balanced after acquisition with Adobe Photoshop).
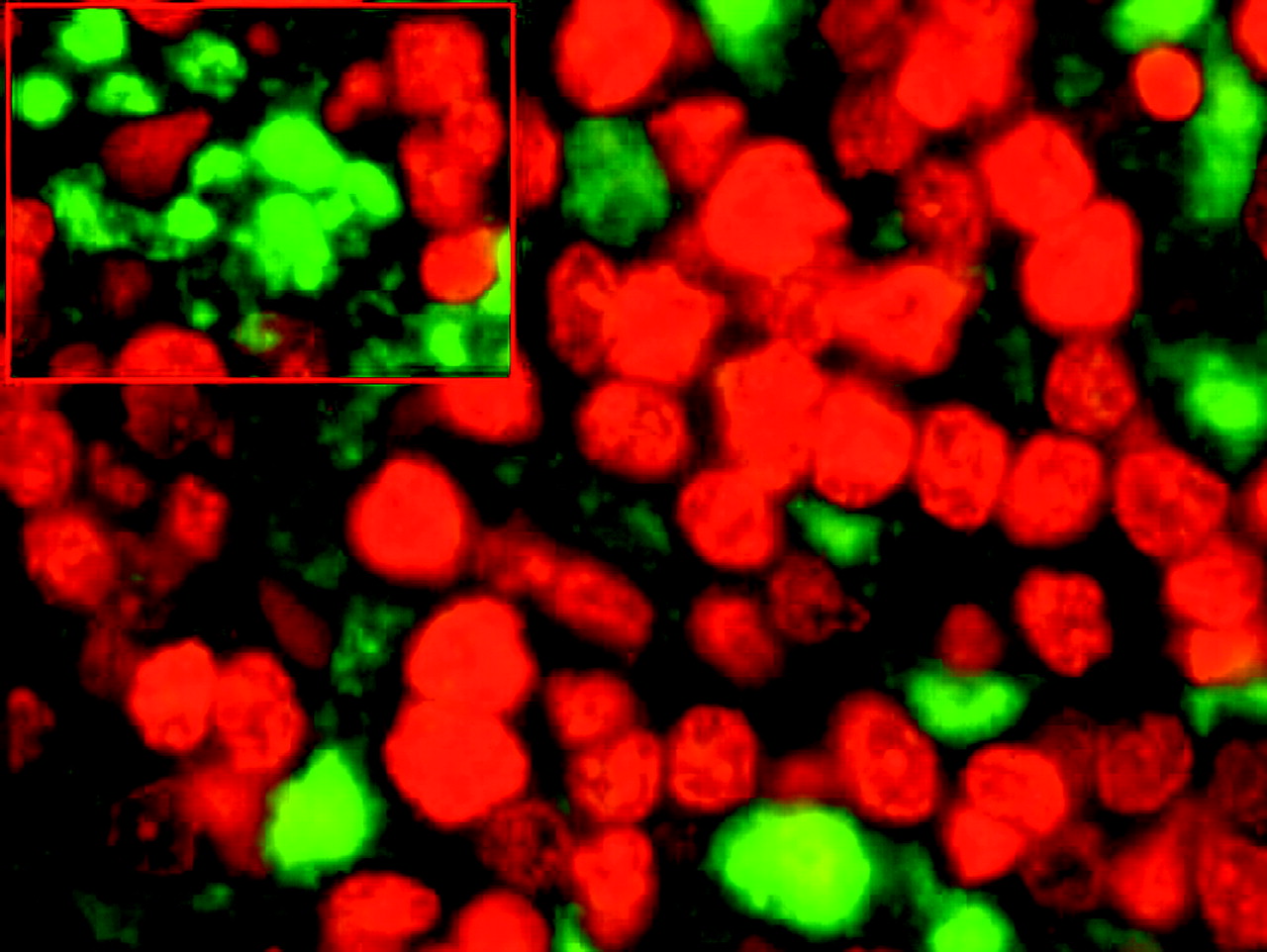
CDKN1B/p27 and Ki67 double immunostaining in a peripheral T cell lymphoma not otherwise specified sample. Note: CDKN1B/p27 as green fluorescent signals and Ki67 as red fluorescent signals (enlarged in the inset). DAPI (4′,6-diamidino-2-phenylindole) staining was adopted as a counterstain (Olympus BX61 microscope, Olympus DP70 camera; CellIF digital imaging solution; Olympus Italia Srl, 20090 Segrate (Milano) Italia).
Direct sequencing
As genomic structural abnormalities may affect CDKN1B/p27 function, we also studied its genomic locus in 81 PTCL/NOS (including those studied by GEP and IHC). Direct sequencing of CDKN1B coding and promoter regions was then used to evaluate the possible existence of mutations, deletions or possible SNPs known to affect protein viability.16 27 28 This approach underlined the presence of two SNPs in the CDKN1B sequence: one (−79C/T) localised in the promoter region, and the other (326T/G) localised in the coding sequence. Specifically, the SNP (−79C/T) was found in 22/81 cases (9/81 were not able to be evaluated). However, this SNP had not been previously associated with significant functional consequences and, in our series, was not associated with increased/reduced CDKN1B expression. On the other hand, as far as SNP 326T/G was concerned, 28/81 PTCLs/NOS turned out to be TT homozygous, 45/81 were heterozygous (TG) and 8/81 were GG homozygous. This SNP has been reported to determine functional consequences, by causing a glycine to valine substitution at codon 109 in TT homozygous cases.29 Noteworthy, the valine variant was associated with decreased levels of p27. In particular, the presence of valine allele was shown to be able to modify the binding between p27 and its negative regulators, by supporting p27 degradation. Furthermore, such a phenotype was associated with a more aggressive tumour stage and possibly poor prognosis in several different cancers.27 28 We thus compared CDKN1B expression at the mRNA level and p27 expression with IHC in cases with and without such polymorphism. Nevertheless, we did not find any significant difference. Of note, such cases did not present with higher Ki-67 expression (as possible indicator of reduced p27 activity) as well. Similarly, we found the CDKN1B locus to be intact in the course of a high-density karyotyping study carried out by the Affymetrix 250k SNPs arrays in a series of PTCLs/NOS.30 Overall, the CDKN1B gene did not appear to be affected by structural abnormalities in PTCL/NOS.
Survival analysis
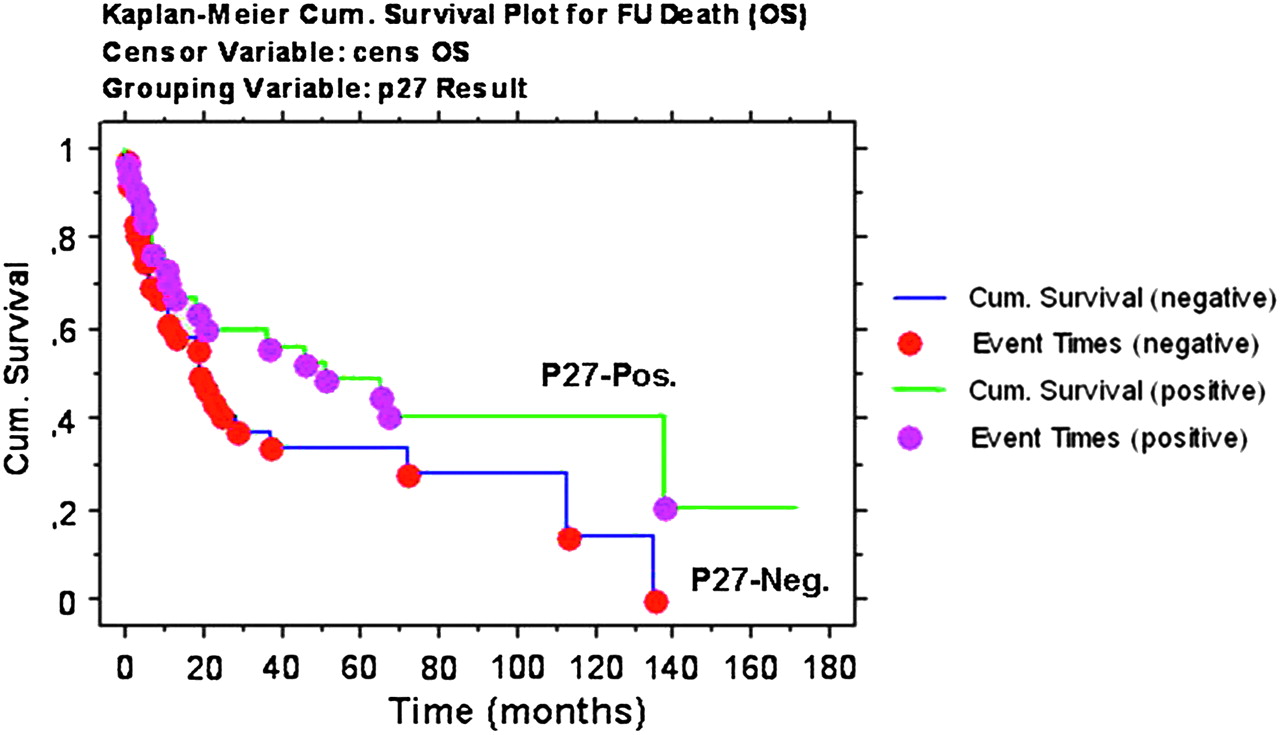
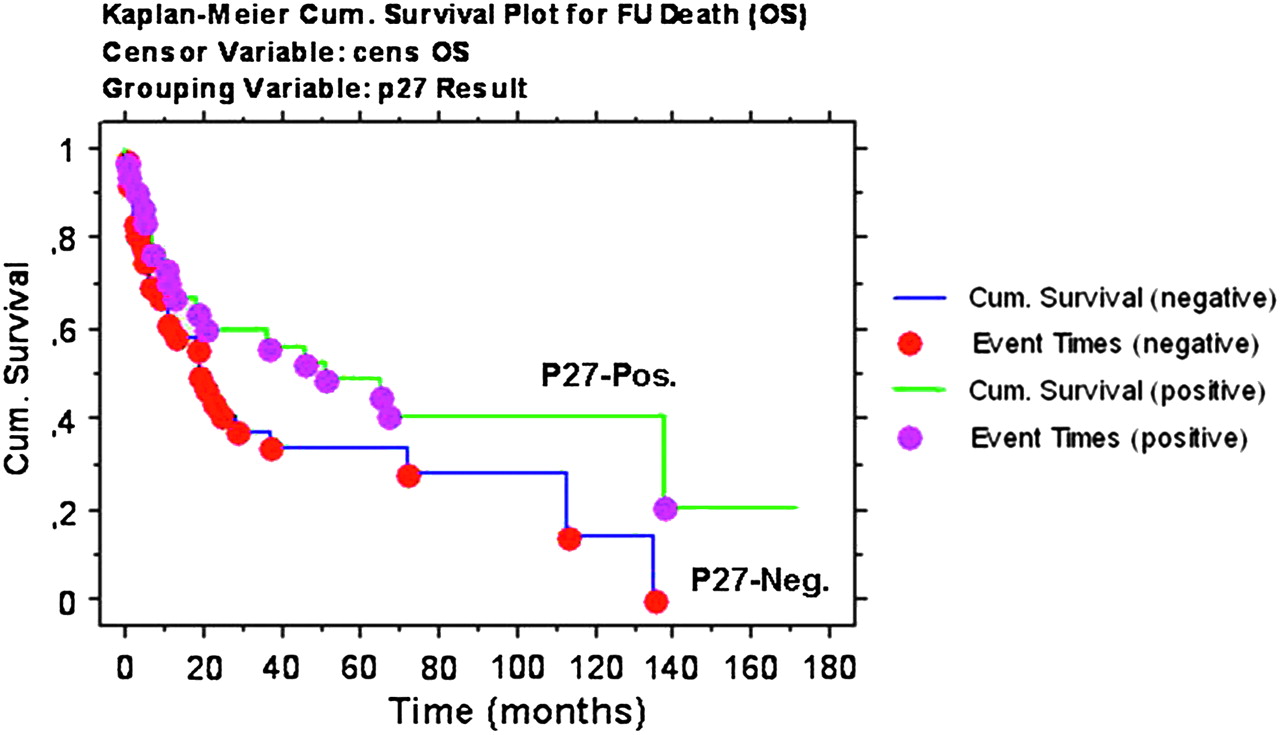
Finally, we investigated the possible prognostic impact of p27 expression, by analysing the 67/98 cases of the TMAs for which complete follow-up was available. However, we failed to detect any significant difference in overall survival in patients with or without p27 expression (p=0.1; figure 4). Similarly, Cox regression did not show any correlation between p27 expression (evaluated as percentage of stained cells) and overall survival. Finally, no survival differences were recorded among patients carrying different CDKN1B SNPs (p=0.4; not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Survival analysis. Overall survival curves obtained by the Kaplan–Meier method. No significant differences were observed for p27-positive and p27-negative cases (log rank p=0.1).
Discussion
In the recent years, several new insights have been provided into the pathobiology of PTCL/NOS.2 Indeed, gene expression profiling has indicated cell proliferation among the most affected cellular functions in this tumour.5 Notably, the proliferation index was also a relevant prognostic feature in independent series.6 7 In this study, we investigated the possible occurrence of CDKN1B/p27 aberrations in PTCL/NOS, as it is frequently responsible for proliferation abnormalities in human cancer. Notably, p27-deficient mice demonstrated multiple defects in growth control but were not particularly inclined to developing cancer.31 32 In particular, though some of the mice developed pituitary adenomas, spontaneous aggressive malignancies were rare. However, p27-deficient mice developed aggressive malignancies when other genomic abnormalities were present.25 In this regard, it has been suggested recently that p27 loss may contribute to T cell lymphomagenesis, based on animal models.25 In particular, it has been hypothesised that p27 deficiency cooperates with BCL2 in tumour formation. Consistently, lymphoma development has been dramatically accelerated by combining p27 loss with Bcl-2 overexpression: p27–/– Lck-Bcl-2 mice develop T cell hyperplasia and spontaneous T cell lymphomas with high penetrance.25 Overall, these data support the concept that that the absence of p27, though not properly oncogenic per se, may facilitate lymphoma development.25 On the other hand, possible p27 alterations have not been exhaustively studied in the field of human PTCLs, as a unique report described p27 loss in ALCL.26
In our series, we failed to identify any relevant abnormality at the genomic and protein levels. First, gene expression analysis did show that CDKN1B expression in all studied PTCLs/NOS was not significantly different from that recorded in non-neoplastic lymph nodes and T lymphocytes.
Second, we studied p27 expression by immunohistochemistry and we found a variable degree of expression among PTCL/NOS cases, similar to that observed in control samples. Furthermore, as physiologically Ki67 and p27 expression are mutually exclusive, we investigated whether the two molecules could be co-expressed by neoplastic cells. Interestingly, double-staining immunofluorescence documented that p27 and Ki67 were not co-expressed, as in reactive conditions. However, as genomic abnormalities can alter p27 function, we also evaluated the integrity of the CDKN1B locus by direct sequencing. In fact, mutations of CDKN1B have been associated with p27 overexpression and consequent drug resistant quiescent phenotype and with p27 downregulation and resulting in hyperproliferative aggressive behaviour.8 15 19 Indeed, we did not find mutations or other structural abnormalities or significant SNPs, confirming that there was no primary alteration in p27 function in our tumour series.
Interestingly, in ALCL p27 loss has been reported to be strongly correlated with PTEN loss,26 sustaining the concept that the two molecules often cooperate in malignant transformation. On the other hand, in line with this idea, we recently demonstrated that PTEN abnormalities are indeed exceptional in a large series of PTCL/NOS.33
Finally, as the proliferation rate was associated with clinical outcome in PTCL/NOS patients,6 7 we investigated whether p27 expression was correlated with any clinical parameter or survival. Basically, though a slight tendency in favour of p27+ patients was observed, the difference was not significant, while no clinical parameter was related to p27 levels. This fact probably reflected the high variability of degrees of expression of this molecule among different cases and the existence of other factors underlying the proliferative alteration in PTCLs.
In conclusion, our study, which is believed to be the first to focus on CDKN1B/p27 expression in PTCL/NOS, provided various pieces of evidence that, taken together, seem to exclude a primary role of CDKN1B/p27 abnormalities in PTCL/NOS pathogenesis.
Take-home messages
Peripheral T cell lymphoma not otherwise specified (PTCL/NOS) usually presents with cell cycle deregulation.
CDKN1B/p27 represents a key regulator of cell cycle progression and is frequently altered in human cancer.
By studying CDKN1B/p27 mRNA expression and protein production, as well as the integrity of the genomic locus, we provided evidence that CDKN1B/p27 aberrations are uncommon in PTCL/NOS.
Acknowledgments
The Authors are grateful to Dr P Artioli, Mr L Chilli, Dr M Piccioli, Miss F Sandri, Dr Gianpaolo Da Pozzo, Dr Fabio Cava and Dr. A. Valdisserri for their skilled technical assistance.
References
Supplementary materials
Web Only Data JCP.2010.083832
Footnotes
Stefano A Pileri and Pier Paolo Piccaluga contributed equally to this work.
Funding This work was supported by Centro Interdipartimentale per la Ricerca sul Cancro ‘G. Prodi’, Bologn AIL, AIRC (IG4987 and IG10007), FIRB (Professor Pileri and Professor Zinzani), RFO (Professor Pileri, Professor Zinzani, Dr Piccaluga), Fondazione Cassa di Risparmio in Bologna, Fondazione della Banca del Monte e Ravenna, Progetto Strategico di Ateneo 2006 (Professor Pileri and Dr Piccaluga).
Competing interests None.
Ethics approval This study was conducted with the approval of the ethics committee of S. Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy.
Provenance and peer review Not commissioned; externally peer reviewed.