Article Text

Abstract
The role of Wnt signalling in the serrated neoplastic pathway of colorectal tumourigenesis appears to be heterogeneous. Wnt pathway abnormalities contribute to the progression of at least a subset of traditional serrated adenomas of the colorectum, but may play a less active role in its pathogenesis compared with that in conventional adenoma-carcinoma. However, immunohistochemical studies of β-catenin in sessile serrated adenomas have shown wide variability, producing conflicting results on Wnt signalling activation in sessile serrated adenomas. DNA methylation, involving APC, SFRPs and mutated in colorectal cancer (MCC), may bridge the mutational gap of APC or β-catenin for activating Wnt signalling in serrated adenomas of the colorectum.
- Wnt signalling pathway
- serrated adenoma
- colorectal cancer
- methylation
- colon
- cancer
Statistics from Altmetric.com
Introduction
Most colorectal cancers (CRCs) develop through a conventional adenoma (CA)–carcinoma sequence.1 ,2 However, recent increasing evidence suggests that 10%–20% of CRCs develop through the ‘serrated polyp-neoplasia pathway’.3–6 The recognition of this pathway in recent years has led to a paradigm shift in our understanding of the molecular basis of CRC and significant changes in clinical practice.7
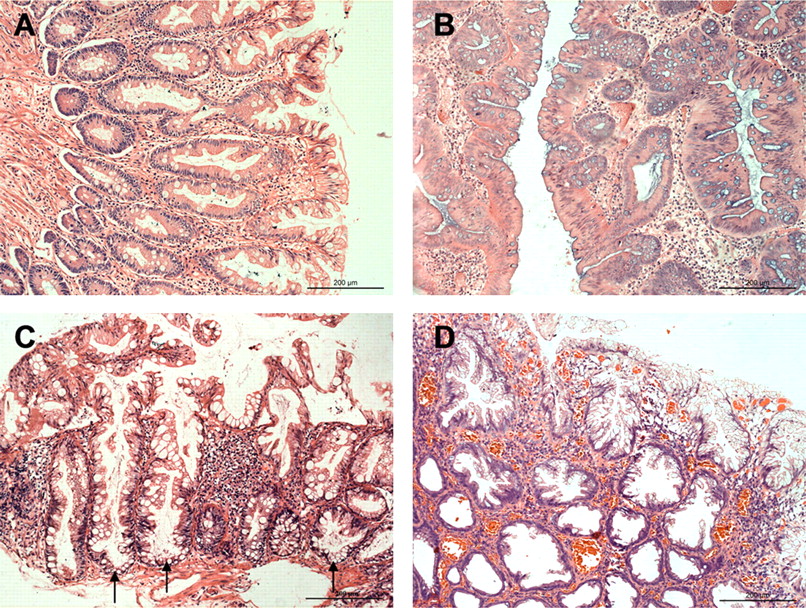
Serrated polyps are histologically classified into hyperplastic polyps (HPs), traditional serrated adenomas (TSAs) and sessile serrated adenomas (SSAs).8 ,9 HPs are characterised by their small size (usually <0.5 cm), a predominantly left-sided location and generally regarded as non-neoplastic. Histologically, HPs have prominent serrations towards the luminal surface and crypts that narrow towards the muscularis mucosae (figure 1A). HPs are further classified as microvesicular HP, goblet cell HP, and mucin-depleted HP.10 Some data suggest that MVHP (especially right-sided) may be a precursor to more advanced SSAs.8 ,11 ,12

Histological features of serrated polyps. (A) Hyperplastic polyp: The serrations of the epithelium are confined to the upper half of the crypts, and the numbers of goblet cells are decreased relative to the normal mucosa. (B) Traditional serrated adenoma: This exophytic polyp shows serrated epithelial changes in association with cytoplasmic eosinophilia, nuclear enlargement and crowding. (C,D) Sessile serrated adenoma: In contrast to the hyperplastic polyp shown in A, the architecture of the crypts is distorted, seen as basal crypt dilatation (arrows) and crypt branching. H&E staining.
TSAs were first described by Longacre and Fenoglio-Preiser13 in 1990 as serrated adenomas (SAs) that exhibit cytological dysplasia reminiscent of classical adenomas and a serrated architecture resembling HPs. TSAs are characterised by a predilection for the distal colon and rectum, apparent dysplasia, nuclear stratification and penicillate nuclei with abundant to moderate eosinophilic cytoplasm (figure 1B). Although TSAs were considered to be lesions of minor importance because of its low prevalence (2%–3.5%),7 recent reports claimed that TSAs have a higher growth rate than CAs, and that the subsequent cancer risk rate at least equals that of CAs.14 ,15
In 1996, the term sessile serrated adenoma was introduced to refer to a subset of serrated lesions that cytologically resemble HPs but is distinguished from HPs on the basis of crypt dilation, branching and horizontal spreading (figure 1C,D).16 Unlike HPs, SSAs are characterised by a larger size, a predominantly right-sided location and the neoplastic potential. However, histopathological diagnosis of SSAs may be difficult, as they closely resemble HPs.17 ,18 The concept of SSAs was reintroduced in 2003 and were accounted to represent 18%–22% of serrated polyps.8 ,19 With the growing evidence of malignant change of SSA in epidemiological backgrounds, SSA has been considered to be the precursor of some microsatellite instability (MSI)-high carcinomas of the proximal colon.9 Some researchers concluded that SSAs were high-risk lesions, with 15% of the SSA patients developing subsequent CRCs or adenomas with high-grade dysplasia.20 Furthermore, there is evidence that the neoplastic progression within this pathway is faster than within the classical adenoma–carcinoma sequence.14 ,21 However, the details of the molecular mechanism of stepwise progression from SSA to early invasive carcinoma remain unclear.
It is well known that Wnt signalling pathway involving β-catenin plays a critical role in colorectal carcinogenesis (figure 2).22 ,23 β-Catenin and its regulators, such as adenomatous polyposis coli (APC), is well studied in CAs and related carcinomas. However, the published data on Wnt pathway activation in SAs (including TSAs and SSAs) are controversial.24–28

{kind=link}
{kind=link}
A canonical Wnt signalling pathway. In the absence of Wnt signal, β-catenin is recruited into the APC/Axin/GSK3β/CK destruction complex and phosphorylated by GSK3β. The hyperphosphorylated β-catenin is targeted for degradation by the proteosome. Binding of Wnt ligand to a Frizzled/LRP-5/6 receptor complex, leading to the inhibition of APC/Axin/GSK3β/CK-mediated β-catenin degradation. Stabilised β-catenin forms a transcriptional complex with Tcf/lef and activates downstream targets such as c-myc.
The purpose of this review is to discuss the possible role and regulatory mechanisms of Wnt signalling pathway in the development and progression of serrated polyps.
Wnt signalling pathway may play a less active role in the pathogenesis of a subset of TSAs
Activation of the Wnt signalling pathway in CRC occurs almost invariably through mutation of the two key molecules: APC and β-catenin (CTNNB1).29 Up to 80% of CRCs have APC mutations, whereas 10% of CRCs have β-catenin mutations.29 An important function of the APC protein is to form a complex with the key effector β-catenin. Loss of APC function leads to translocation of β-catenin from the lateral cell membrane to the nucleus, where it promotes the transcription of multiple genes involved in tumour growth and invasion.30 Although aberrant activation of Wnt signalling is a prerequisite for the initiation of most CRCs,29 its role in TSAs is still controversial.
Some studies report a high incidence of APC gene mutation/ loss of heterozygosity (LOH) in TSAs. Hiyama and colleagues25 demonstrated APC mutation in 40% (2/5) of TSAs. In Matsumoto's study,31 3/3 TSAs with familial adenomatous polyposis had germline APC mutation. In another study, LOH of APC was seen in 20% (7/35) of TSAs.32 However, most of these studies were limited by their small sample sizes.
Not all molecular studies have shown consistent findings. Sawyer et al 33 reported that APC mutations occurred in 12.8% (5/39) of TSAs. In a study by Dehari et al,27 APC mutations were detected in 3.8% (1/26) of TSAs, and in none of the four adenocarcinomas with TSAs, but were detected in 66.7% (8/12) of tubular adenomas and in 50% (4/8) of adenocarcinomas with tubular adenomas. Uchida and colleagues26 found that 37.0% (10/27) of CAs showed somatic mutations of APC but no APC mutation was identified in 12 TSAs. In our previous study,34 the frequency of APC gene mutation in TSAs (8.3%, 1/12)was significantly lower than that in CAs (56.3%, 9/16) and CRCs (50.0%, 7/14). These studies looking for APC mutations in TSAs have shown a low rate of detection, relative to the 50%–63% reported in CAs.35
Sawyer et al 33 argued that APC mutations in these studies were likely to be underestimated because of poor quality of DNA retrieved from the archival material. However, we noticed that most of these studies had DNA from paraffin tissue sections of CAs as controls, which showed a high frequency of APC mutations. Moreover, even 3/3 TSAs had germline APC mutations in the study by Matsumoto et al,31 these mutations had been shown to cluster at the proximal exons 9 and at the distal 3′end of exon 15. It is reported that these mutations do not significantly affect the tumour-suppressor function of APC,36 ,37 suggesting that TSA of familial adenomatous polyposis may be a phenotype associated with less impaired APC function, although this study was limited by its very small sample size. Indeed, our previous study found that expression of APC protein in TSAs was higher than that in CAs, although the difference was not statistically significant.34 These findings suggest that APC's function in TSAs is less likely to be impaired compared with that in CA–carcinoma sequence.
Previously published results of β-catenin expression in TSAs had shown mixed findings. For instance, in a study by Jiao et al 24 in 2008, nuclear β-catenin positive cells were found in all TSAs (43 cases), but there was no significant difference in nuclear β-catenin positive rates between TSAs (30%) and CAs (30%).24 In another study, Oh et al 38 also noted a relatively high frequency of positivity for β-catenin in TSAs (61%, 14/23).
However, contradictory results have been reported by other investigators. Sawyer et al 33 evaluated β-catenin expression in 39 TSAs and found that only 12.8% (5/39) of TSAs showed nuclear β-catenin staining and 1/39 showed β-catenin mutation. Yamamoto et al 28 reported that widespread or focal nuclear β-catenin expression was demonstrated in 7% of TSAs (3/45), relative to 61% of CAs (43/71). Cytoplasmic immunostaining for β-catenin was demonstrated in 16% of TSAs (7/45) relative to 77% of CAs (55/71). No mutation in exon 3 of β-catenin was found in TSAs, while 7% of CAs (5/71) had β-catenin mutations. On the basis of these results, Yamamoto et al 28 concluded that intracellular localisation of β-catenin may not be associated with an early event of the tumour progression in most TSAs. However, Yachida et al 39 reviewed the immunostaining results presented in that paper, and argued that the figure indicated scattered positive nuclear labelling (not negative staining, as the authors pointed out).
In our previous study in 2009,34 nuclear expression of β-catenin was not seen in any of the TSAs (12 cases). Cytoplasmic accumulation of β-catenin was demonstrated in 41.7% (5/12) of TSAs, relative to 68.6% (11/16) of CAs. No β-catenin mutation was detected in any of the 12 TSAs. In our more recent study in 2011,40 nuclear labelling of β-catenin was detected in 19.6% of CAs (51 cases), but in none of the TSAs (37cases). Cytoplasmic accumulation of β-catenin was found in 21.6% of TSAs, significantly lower than that in CAs (60.8%).
Although conflicting results exist, Wnt pathway abnormalities (involving changes in APC/β-catenin) may contribute to the progression of at least a subset of TSAs of the colorectum. However, it is worth noting that there seems to be a trend that frequency of Wnt signalling abnormality in TSAs is lower than that in CAs, raising the possibility that the Wnt pathway may play a less active role in the pathogenesis of a certain proportion of TSAs.
Conflicting results on activation of the Wnt signalling pathway in SSAs
The published data on Wnt pathway activation in SSAs are more conflicting. In a study by Wu et al,41 the authors surprisingly found that predominant nuclear labelling for β-catenin was noted in 9/22 SSAs (41%) but in none of the HPs. In all SSAs with nuclear labelling, membranous labelling was significantly decreased. They expanded upon this observation of β-catenin expression in a larger set of serrated polyps of the colorectum.39 Immunolabelling for β-catenin confirmed the presence of nuclear accumulation in 35/54 (67%) SSAs, compared with 0/12 HPs. Furthermore, nuclear β-catenin labelling was seen in 8/27 (29%) SSAs without dysplasia, but in 27/27 (100%) SSAs with dysplasia. Molecular study revealed that aberrant nuclear labelling of β-catenin occurred in a background of BRAF activation.39 Their findings suggest that aberrant Wnt signalling has a role in the development of a subset of SSAs and nuclear accumulation of β-catenin may be closely related to the early progression of SSAs.
The results from Sandmeier et al 42 showed that abnormal β-catenin expression (loss of membranous β-catenin expression and nuclear β-catenin accumulation) was present in 37.5% (6/16) of SSAs, but not significantly different from 50.0% (6/12) of HPs. By contrast, in a study by Joo et al,43 nuclear β-catenin staining was observed in 0% of SSAs. However, this study was limited by its small sample size (n = 10). In another study by Fujita et al,44 no nuclear β-catenin expression was seen in 53 SSAs, although nuclear accumulation of β-catenin was observed in 50% (6/12) of SSAs with neoplastic progression (SSANs, including intraepithelial high-grade dysplasia and submucosal invasive carcinoma). These results are consistent with our previous study showing that nuclear labelling of β-catenin was detected in none of the SSAs and HPs tested.40 These results indicate that Wnt pathway activation, seen as nuclear labelling for β-catenin, is unlikely to contribute to tumourigenesis in most SSAs.
Remarkably, these immunohistochemical studies of β-catenin in SSAs have shown wide variability in terms of proportion with nuclear staining, from 0% to 100%. Many reasons may contribute to these differing results, including intrinsic tumour heterogeneity, different stages of adenomas, small sample size, differences in antigen retrieval, antibodies and staining procedures used by each laboratory and different evaluations of immunostaining with varying degrees of sensitivity. Especially, conclusive studies of the Wnt signalling pathway in SSAs have been limited by the heterogeneity of SSAs classified in this category. Diagnostic criteria and nomenclature for these lesions are inconsistent and partly confusing.18 ,45 Therefore, some SSAs are still misdiagnosed and wrongly categorised, which may lead to differing results. Fortunately, a standardised diagnostic nomenclature of different serrated polyps was formulated in 2010,45 meriting future research and clinical management of serrated polyps.
Epigenetic mechanisms in activating Wnt signalling of the serrated neoplastic pathway
Activating mutations in CTNNB1 and APC are a common feature of CA–carcinoma sequence. Up to 80% of CRCs have APC mutations, whereas 10% of CRCs have β-catenin mutations.29 However, most studies reported infrequent APC mutations in TSAs compared with that in CAs.26 ,27 ,33 ,34 MSI is classically a feature of TSAs that appear to distinguish them from CAs.33 ,46 ,47 It is reported that activating mutations in CTNNB1, not APC genetic inactivation, are frequent features of MSI CRCs that similarly cause stabilisation of β-catenin.48 ,49 However, activating mutations in CTNNB1 is seldom seen in TSAs.28 ,33 ,34 Yachida and colleagues39 also found no mutations in CTNNB1 as a mechanism for Wnt activation, although they noted frequent β-catenin nuclear labelling in SSAs.
These results suggest that nuclear accumulation of β-catenin may also occur in the absence of mutations in the APC/β-catenin pathway.33 Previous studies have revealed that DNA methylation could provide an alternative mechanism to gene mutation for silencing APC. 50–52 Moreover, Suzuki et al 53 provided compelling evidence of the Wnt pathway activation in CRC cell lines by hypermethylation of the Wnt pathway antagonists SFRP1, SFRP2 and SFRP5. Indeed, a high frequency of DNA methylation is common in SAs and may play an important role in their pathogenesis.3 ,54–56 Thus, it is conceivable that DNA methylation could bridge the mutational gap of APC or CTNNB1 for activating Wnt signalling in SAs.39 Dhir et al 57 reported that SFRPs (1, 2, 4 and 5) were methylated frequently in SSAs and SSAs with dysplasia, but infrequently in HPs, providing evidence that the Wnt signalling pathway may be activated by epigenetic mechanisms in SSAs. Our previous study has also shown that hypermethylation of APC promoter 1A, instead of mutations involving APC and β-catenin, contributed to moderate activation of Wnt signalling in a subset of TSAs.34 Whether APC methylation also plays a role in tumourigenesis of SSA remains to be tested.
Recent studies have shown that mutated in colorectal cancer (MCC), a candidate tumour suppressor, can also interact with β-catenin, block β-catenin/TCF/LEF DNA binding, and affect endogenous β-catenin localisation/levels.58 Moreover, MCC methylation, associated with BRAF mutation, was more common in serrated polyps than in CAs.59 Therefore, MCC inactivation by promoter methylation could be an initiating Wnt pathway-activating event in the serrated neoplasia pathway. Additional studies will be required to address this possibility.
Take-home messages
-
Wnt pathway abnormalities contribute to the progression of a subset of TSAs, but may play a less active role compared to that in conventional adenoma-carcinoma sequence.
-
The role of Wnt signaling in the progression of SSA is still controversial. Further studies are needed to clarify this issue.
-
DNA methylation may bridge the mutational gap for activating Wnt signaling in the “serrated polyp-neoplasia pathway” of the colorectum.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.