Article Text

Abstract
Hyperparathyroidism is a common endocrine disorder with potential complications on the skeletal, renal, neurocognitive and cardiovascular systems. While most cases (95%) occur sporadically, about 5% are associated with a hereditary syndrome: multiple endocrine neoplasia syndromes (MEN-1, MEN-2A, MEN-4), hyperparathyroidism-jaw tumour syndrome (HPT-JT), familial hypocalciuric hypercalcaemia (FHH-1, FHH-2, FHH-3), familial hypercalciuric hypercalcaemia, neonatal severe hyperparathyroidism and isolated familial hyperparathyroidism. Recently, molecular mechanisms underlying possible tumour suppressor genes (MEN1, CDC73/HRPT2, CDKIs, APC, SFRPs, GSK3β, RASSF1A, HIC1, RIZ1, WT1, CaSR, GNA11, AP2S1) and proto-oncogenes (CCND1/PRAD1, RET, ZFX, CTNNB1, EZH2) have been uncovered in the pathogenesis of hyperparathyroidism. While bi-allelic inactivation of CDC73/HRPT2 seems unique to parathyroid malignancy, aberrant activation of cyclin D1 and Wnt/β-catenin signalling has been reported in benign and malignant parathyroid tumours. Clinicopathological correlates of primary hyperparathyroidism include parathyroid adenoma (80–85%), hyperplasia (10–15%) and carcinoma (<1–5%). Secondary hyperparathyroidism generally presents with diffuse parathyroid hyperplasia, whereas tertiary hyperparathyroidism reflects the emergence of autonomous parathyroid hormone (PTH)-producing neoplasm(s) from secondary parathyroid hyperplasia. Surgical resection of abnormal parathyroid tissue remains the only curative treatment in primary hyperparathyroidism, and parathyroidectomy specimens are frequently encountered in this setting. Clinical and biochemical features, including intraoperative PTH levels, number, weight and size of the affected parathyroid gland(s), are crucial parameters to consider when rendering an accurate diagnosis of parathyroid proliferations. This review provides an update on the expanding knowledge of hyperparathyroidism and highlights the clinicopathological correlations of this prevalent disease.
- PARATHYROID
- GENETICS
- ENDOCRINE PATHOLOGY
Statistics from Altmetric.com
Introduction
Hyperparathyroidism (HPT) is a common endocrine disorder characterised by oversecretion of parathyroid hormone (PTH), which may be autonomous (independent of serum calcium levels) or the result of a physiological stimulus.1–11 Our understanding of this disease has progressed substantially over the past decade, with advancements made in biochemical, radiological and molecular testing.1 ,3 ,12 ,13 Early detection is crucial to prevent complications from uncontrolled HPT, affecting the kidneys (ie, nephrolithiasis or nephrocalcinosis) and bones (ie, osteoporosis, osteitis fibrosa cystica).4 ,6 ,7 ,14–19 Furthermore, neurocognitive dysfunction and cardiovascular morbidity and mortality appear to be increased in patients with overt HPT, although the nature and reversibility of these effects remains less evident.4 ,14 ,15 ,20–25
HPT is generally divided into three types: primary (autonomous HPT), secondary (HPT that results from a chronic stimulus causing PTH secretion) and tertiary (emergence of autonomous HPT in refractory secondary HPT).1–9 Primary HPT is currently the most common cause of hypercalcaemia, with an annual incidence ranging from 34 to 120 cases per 100 000.4 ,7 ,14 ,26–28 While most cases are sporadic (95%), about 5% present with a hereditary syndrome: multiple endocrine neoplasia syndromes (MEN-1, MEN-4, MEN-2A), hyperparathyroidism-jaw tumour syndrome (HPT-JT), familial hypocalciuric hypercalcaemia (FHH-1, FHH-2, FHH-3), familial hypercalciuric hypercalcaemia, neonatal severe HPT and isolated familial HPT.4 ,7 ,29–33
Surgical resection of abnormal ‘hyperfunctioning’ parathyroid gland(s) remains the only curative treatment for primary HPT.1 ,4 ,7 ,29 ,34–37 Thus, parathyroidectomy specimens are frequently encountered in this setting.3 ,38–45 Clinicopathological correlates of primary HPT include parathyroid adenoma (80–85%), hyperplasia (10–15%) and carcinoma (<1–5%).4 ,7 ,38 ,39 ,41 ,42 ,44 ,46–49 Secondary HPT typically presents with diffuse parathyroid hyperplasia, whereas tertiary HPT reflects the emergence of autonomous PTH-producing adenoma or rarely carcinoma from a background of secondary hyperplasia.7 ,11 ,42 ,44 ,46 Accurate subtyping is important for treatment decision-making and requires a thorough integration of pathological features with clinical, biochemical, radiological and intraoperative findings.1 ,4 ,7 ,42 ,49–51 This review provides an update on the evolving knowledge of HPT and highlights the clinicopathological correlations of this important disease.
Clinical and biochemical features
Clinically, sporadic primary HPT can present at any age, with an increased incidence in postmenopausal women.4 ,7 ,30 ,42 ,52 ,53 A wide spectrum of clinical manifestations can be encountered, depending on the duration and degree of PTH oversecretion and resultant hypercalcaemia (tables 1 and 2).4 ,6 ,7 ,12 ,14 In the past, symptomatic disease was the most common presentation, prompting the ‘classic’ description of ‘stones’ (ie, nephrolithiasis), ‘bones’ (ie, osteitis fibrosis cystica) and ‘groans’ (ie, constipation, ileus).4 ,6 ,12 ,14 ,15 However, with the advent of routine serum calcium testing, most cases (70–80%) are now diagnosed incidentally, in the ‘asymptomatic’ form.7 ,12 ,14 The natural progression from asymptomatic to symptomatic disease remains unclear at this time.4 ,7 ,12 ,14 However, even its ‘asymptomatic’ form, HPT is associated with potential morbidities, including decreased trabecular/cortical bone density and clinically ‘silent’ nephrolithiasis/nephrocalcinosis, which can be detected radiographically.6 ,12 ,14 ,16–19 ,54–62 In light of these findings, all cases of incidentally detected hypercalcaemia merit additional investigations, including ‘intact’ serum PTH measurement, to clarify its aetiology (PTH dependent vs PTH independent).4 ,6 ,7 Other commonly described symptoms in HPT are associated with hypercalcaemia (ie, weakness, easy fatigability, anxiety, cognitive impairment).4 ,7 ,12 ,14 ,15 ,20 ,54 Gastrointestinal manifestations are almost never reported in the modern presentation of primary HPT.6 ,14 ,15 ,63 ,64
Brief overview of hyperparathyroidism
Clinicopathological correlates of primary hyperparathyroidism
Biochemically, mild hypercalcaemia (usually within 0.25 mmol/L of normal range) is often the first finding in sporadic primary HPT.4 ,6 ,7 ,65 Typically, urinary calciuria is normal or increased although it may be low in individuals with concomitant thiazide or lithium use.66 ,67 A persistently low calciuria after thiazide or lithium discontinuation should raise the possibility of FHH. A normocalcemic variant of primary HPT, defined as high serum PTH and normal serum ionised calcium in the absence of known causes of secondary HPT, has been recently described.2 ,4 ,6 ,7 ,12 ,14 ,24 ,68–70 The epidemiology, natural history and management of normocalcemic primary HPT remain unclear at this time; however, undoubtedly a subset of individuals will develop hypercalcaemia, while some show target organ alterations such as decreased bone mineral density.1 ,2 ,4 ,24 ,69 ,71 Secondary HPT presents with high serum PTH and normocalcemia or hypocalcaemia.7 ,10 ,11 ,72 ,73 Although it is initially and adaptive response to a variety of stimuli resulting mainly in extracellular hypocalcaemia (vitamin D deficiency, chronic kidney disease, idiopathic hypercalciuria, calcium malabsorption), long-standing secondary HPT may eventually develop into autonomous HPT (ie, tertiary HPT).7 ,10 ,11 ,42 ,44 ,72 ,74 Similar to primary HPT, tertiary HPT is biochemically characterised by an elevation of both PTH and calcium in the serum.11 ,74 However, tertiary HPT is usually easily distinguished from primary HPT by a readily identifiable aetiology, which in most circumstances is advanced chronic kidney disease.11 In light of these findings, careful assessment of medication history (ie, lithium, thiazide), renal function, urinary calcium and family history is important to exclude secondary/tertiary and hereditary HPT, as their prognosis and management can differ significantly from that of sporadic primary HPT.4 ,6 ,7 ,11 ,42 Serum 25-hydroxyvitamin D levels should also be measured; not only is vitamin D deficiency a frequent cause of secondary HPT, it is commonly associated with primary HPT and can mask the patient's hypercalcaemia.4 ,6 ,7 ,13
Familial syndromes are reported in about 5% of patients with primary HPT.4 ,7 ,13 ,29–32 ,42 ,44 ,75–81 Clinical findings suggestive of hereditary HPT include early onset of disease (<30 years); family history of hypercalcaemia; skin lesions (lipomas, facial angiofibromas, truncal collagenomas), pituitary adenomas and neuroendocrine tumours (including pheochromocytomas) associated with multiple endocrine neoplasia syndromes; jaw tumour with or without renal cysts (HPT-JT syndrome); hypercalcaemia associated with hypocalciuria or a calcium-to-creatinine clearance ratio <0.01 (FHH).4 ,7 ,30–32 ,78 ,82–85 When familial disease is suspected, serum calcium measurement of first-degree relatives should be considered along with other clinical investigations.4 ,6 ,7 ,13 ,30 In cases where MEN-1 is suspected on the basis of multiglandular primary HPT with or without other concomitant MEN-1 associated tumours, testing for a MEN1 mutation is recommended.86 A definite diagnosis of FHH may ultimately require demonstration of a germline mutation in CASR, GNA11 and AP2S1.4 ,13 ,30 ,32 ,42 ,77 ,79 ,87–91 A severe neonatal phenotype (neonatal severe HPT) has also been described, presenting with severe HPT and life-threatening hypercalcaemia in the first six months of life.4 ,5 ,7 ,42 ,44
Although rare, parathyroid carcinoma should be considered in all patients with HPT and the following features: severe hypercalcaemia (albumin-corrected calcium >3 mmol/L), extremely high PTH levels (>3 times the upper limit of normal), concomitant bone and kidney involvement, palpable neck mass, jaw tumour, and family history of parathyroid or other endocrine disorders.4 ,7 ,47 ,49 ,51 ,92–101 Clinically, parathyroid carcinomas tend to present at an earlier age, with equal frequencies in both sexes.42 ,47 ,49 ,51 ,96 ,102 Biochemically, a third-generation/second-generation PTH ratio >1 has been shown to help predict parathyroid malignancy prior to surgery by correlating with other clinical, biochemical and radiological findings.47 ,51 ,103–105 Although this method has not been widely accepted, it has been proposed based on the fact that most parathyroid carcinomas overproduce amino-PTH, which is recognised by third-generation but not second-generation PTH assays.47 ,51 ,81 ,103–105 However, rare ‘non-functioning’ parathyroid carcinomas have also been reported.42 ,47 Radiographic lesions suspicious for distant metastasis may also be identified on sestamibi scintigraphy or CT scan, which can be confirmed on biopsy.4 ,7 ,42 ,47 ,51
Radiological investigations
In conjunction with clinical and biochemical findings, radiological investigations are necessary to guide treatment decision-making in primary HPT (tables 1 and 2).4 ,6 ,7 ,29 ,42 ,97 ,106–108
Extra-parathyroid imaging is performed to assess PTH-related complications on skeletal and renal structures, as positive findings may warrant surgery even in patients with asymptomatic primary HPT.1 ,4 ,29 For instance, renal ultrasonography is performed to exclude clinically ‘silent’ nephrocalcinosis and nephrolithiasis, which can occur in up to 20% of cases.1 ,4 ,7 ,12 ,29 Measurement of bone mineral density at the lumbar spine, hip and distal third of the forearm should also be investigated using dual-energy X-ray absorptiometry (figure 1A).1 ,4 ,7 ,12 ,29 The distal third of the forearm, enriched in cortical bone, is generally the first site to be affected because PTH preferentially affects cortical bone.1 ,4 ,6 ,7 ,12 ,29 The lumbar spine (richer in trabecular bone) is usually more preserved, whereas the hip region (mixture of cortical/trabecular bone) tends to show intermediate findings.1 ,4 ,6 ,7 ,12 ,29 However, it should be noted that a subset of patients (∼15%) present with a different densitometry profile, with mainly vertebral osteopenia or osteoporosis.6 ,12 ,29 ,109 In patients with severe HPT, plain-film radiography and CT imaging may identify signs of bone disease such as pathological fractures, ‘salt and pepper’ appearance of the skull, tapering of the distal third of the clavicles, lytic lesions (figure 1B) of the pelvis, long bones and shoulders, and subperiosteal bone erosions in the distal phalanges.1 ,4 ,6 ,7 ,12 ,16 ,29 ,65 ,110
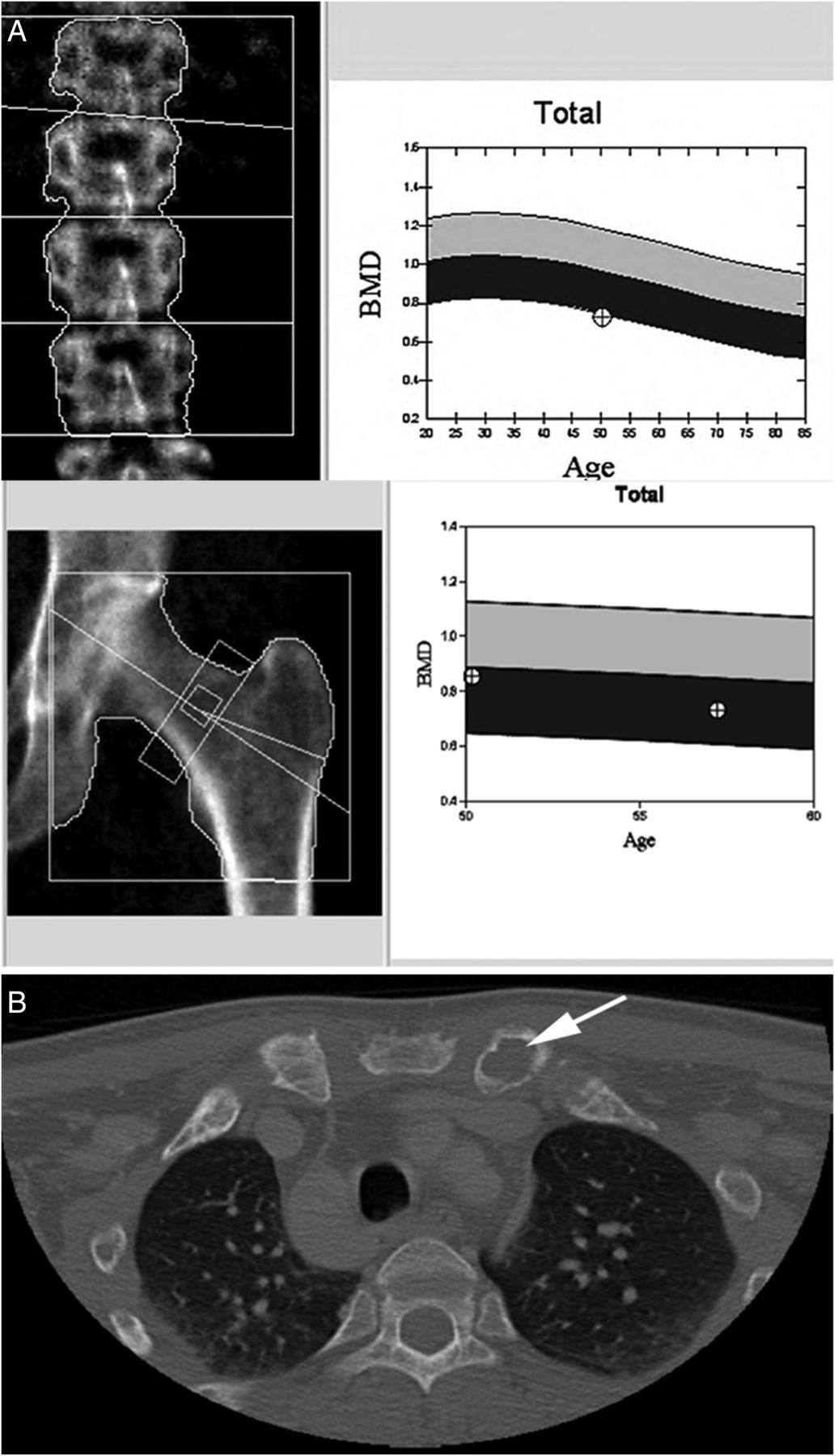
Bone manifestations of primary hyperparathyroidism. (A) Progressive decrease in bone mineral density (BMD) at the lumber spine (−17.1%) and hip (−14.4%) in a 58-year-old woman with primary hyperparathyroidism. (B) CT showing lytic expansile lesion of the left clavicular head in a 30-year-old man with severe hyperparathyroidism.
Parathyroid imaging is used to localise and characterise parathyroid abnormalities preoperatively.4 ,6 ,7 ,51 ,106–108 ,111–113 It is particularly helpful in the identification and characterisation of single-gland disease (adenoma or rare carcinoma) although it may also detect multigland parathyroid disease (usually hyperplasia) allowing for the appropriate surgical approach.4 ,6 ,7 ,51 ,106–108 ,111 ,112 ,114 ,115 In patients with a predisposition for multigland disease (eg, those with MEN), parathyroid imaging is perhaps most useful in the localisation of recurrent disease as their initial surgery usually entails a four gland parathyroid exploration with resection of most of the parathyroid tissue.116 ,117 Although several imaging modalities have been described, neck ultrasound and 99technetium-labelled sestamibi (99mTc-sestamibi) scintigraphy are currently the most commonly used radiological tools (figure 2).4 ,6 ,7 ,51 ,106–108 ,111 ,112 99mTc-sestamibi is taken up by both parathyroid and thyroid tissue, and its uptake is generally increased and prolonged in functioning neoplastic or hyperplastic parathyroid gland(s) (figure 2).4 ,6 ,7 ,51 ,106–108 ,111 ,112 In addition, it has the advantage of localising ectopic parathyroid tissue.4 ,5 ,6 ,7 ,51 ,106–108 ,111 ,112 Recently, dual-tracer subtraction using 99mTc-sestamibi in combination with radioactive iodine (123I) has also been proposed to separate parathyroid uptake of sestamibi from thyroid uptake to achieve better visualisation of the functioning parathyroid tissue.4 ,6 ,7 ,51 ,106–108 ,111–119
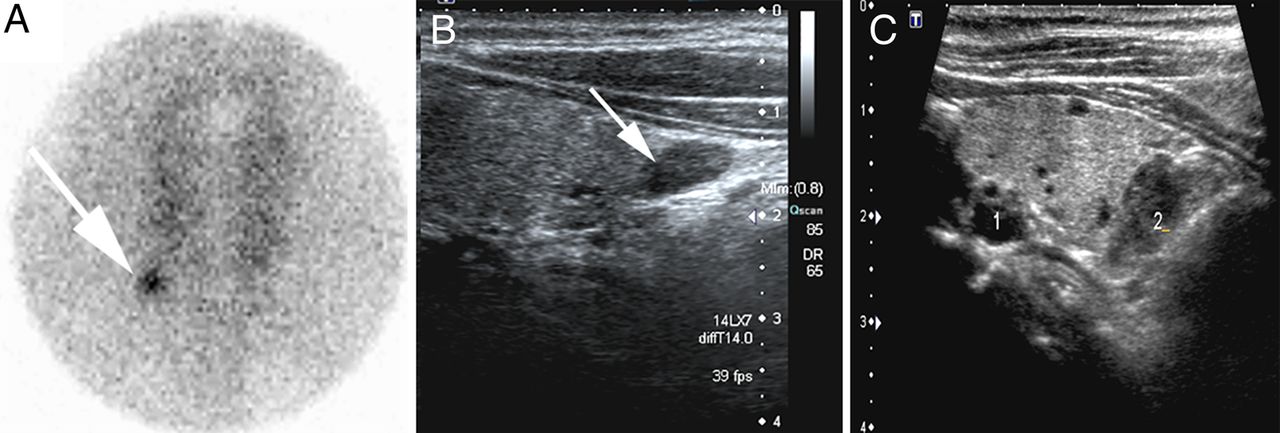
Parathyroid imaging in hyperparathyroidism. Neck ultrasound and 99technetium-labelled sestamibi (99mTc-sestamibi) scintigraphy are currently the most commonly used radiological tools. (A) Focal scintigraphic activity in the inferior right pole of the thyroid corresponding to a right lower parathyroid adenoma. (B) Neck ultrasound showing a hypoechoic nodule in the area of focal 99mTc-sestamibi persistent activity. (C) Ultrasound image shows two hypoechoic nodules behind the left thyroid lobe corresponding to enlarged parathyroids in a patient with secondary hyperparathyroidism due to end stage chronic renal failure.
Pathological manifestations of hyperparathyroidism
In order to better understand the pathological manifestations of HPT (tables 1⇑–3), the characteristics of normal parathyroid glands will also be highlighted here.
Histopathological and molecular features of primary hyperparathyroidism
Normal parathyroid
In healthy individuals, the parathyroid glands are generally 4 in number (two superior, two inferior), although supernumerary parathyroid glands can occur in 2–13% of the population.38 ,42 ,44–46 ,120 It should be noted that all superior glands are located within the thyroid gland or in its pseudocapsule.121 Grossly, a normal parathyroid gland appears brown, round-to-ovoid, small (often <6–8 mm) and can weigh up to 40–60 mg each (figure 3).38–40 ,42 ,44–46 ,99 ,120 Occasionally, parathyroid tissue can occur in ectopic locations (near the carotid bifurcation, pericardial sac, mediastinum, retroesophageal space, vagus nerve, angle of the jaw and thyroid gland) as a result of aberrant migration during embryogenesis.38–40 ,42 ,44–46 ,99 ,120 Histologically, each gland is comprised of glandular parenchyma, adipose tissue and an arborising network of blood vessels, generally confined within a thin pseudocapsule.38 ,42 ,44 ,45 The parenchymal cells are composed predominantly of chief cells (main producer of PTH), along with oncocytic cells (rich in mitochondria; also known as oxyphilic cells or oxyphils) and transitional oncocytic cells (representing an intermediate phase between chief cells and oncocytic cells).38–40 ,42 ,44–46 ,99 ,120

Normal parathyroid gland. A normal parathyroid gland appears grossly brown and round-to-ovoid. It measures less than 6–8 mm and weighs up to 40–60 mg each.
Handling of parathyroidectomy specimens and the role of intraoperative consultation
The clinical history, biochemical work-up, as well as number, size and weight of affected gland(s) are important parameters to consider when rendering a clinicopathological diagnosis of parathyroid proliferations. The initial examination should start with a gross inspection. The parathyroid gland(s) should be carefully weighed and measured after removal of the surrounding fat tissue.39 ,41 ,42 ,44 At this stage, it is critical to identify the vascular pole (hilum) before slicing the gland horizontally to this plane.3 ,40 ,41 ,44 This step is particularly important, given the fact that an atrophic rim of normal parathyroid tissue, one of the key histopathological features of a parathyroid adenoma,42 ,44 ,122 ,123 is often located at the vascular pole (hilum) of the gland.
Accurate diagnosis of parathyroid proliferations can be difficult or even impossible at the time of the intraoperative assessment.39 ,40 ,42 ,43 ,44 ,46 ,123 ,124 The primary role of the intraoperative pathological consultation is to confirm the presence of parathyroid tissue in the resected specimen and to exclude other tissues, such as lymph nodes or thymus, which can be mistaken for parathyroid tissue by the surgeon.35 ,39–46 ,123 ,125 In patients with HPT, intraoperative confirmation of glandular abnormality (enlarged cellular gland) is also required (figure 4). In the past, pathologists were also asked to assess parenchymal-to-adipose cell ratio and to perform fat stains (oil red O or Sudan IV stains) to diagnose parathyroid proliferations.44 ,126 The value of these stains remains controversial, with most experts agreeing that this approach is not reliable in distinguishing parathyroid adenoma from hyperplasia. While the evaluation of a second parathyroid gland may be an alternative to fat stains, the recent development and adoption of rapid intraoperative PTH assay appears to be a more precise tool to distinguish adenoma from hyperplasia.29 ,36 ,42 ,127–129 In patients with single-gland disease, removal of a solitary gland results in reduction of PTH levels of >50%, often >75%, within 10 min.4 ,29 ,34 ,36 ,42 ,130 Consequently, many hospitals with high volumes of parathyroid surgeries use rapid intraoperative PTH measurements, immediately before and 10 min after resection.4 ,29 ,34 ,36 ,42 ,130 If a significant drop (>50%) in PTH levels is noted, no further exploration is undertaken.4 ,29 ,34 ,36 ,42 ,130 In this setting, a definite diagnosis of the parathyroid pathology is not required at the time of surgery.4 ,29 ,34 ,36 ,42 ,130 Immediate hypocalcaemia after surgery is frequently seen in patients with single-gland parathyroid disease and should be monitored accordingly.4 ,29 ,34 ,36 ,42 ,130

The role of intraoperative consultation in hyperparathyroidism. The primary role of the intraoperative pathological consultation is to confirm the presence of parathyroid tissue and to exclude other tissues mimicking parathyroid gland. The parenchymal-to-adipose cell ratio is not a reliable tool to distinguish an abnormal gland. In patients with hyperparathyroidism, intraoperative confirmation of an abnormal gland based on the weight (>60 mg) and size (>8 mm) of the gland is also required. Accurate diagnosis of parathyroid proliferations can be difficult or even impossible at the time of the intraoperative consultation. Thus, the term ‘enlarged cellular gland’ should be applied to abnormal gland at the time of intraoperative consultation. The rapid intraoperative parathyroid hormone assay appears to be a more precise tool to distinguish adenoma from hyperplasia at the time of intraoperative consultation.
Histopathological correlates of HPT
Primary HPT can occur hereditarily (∼5%) or sporadically (∼95%) due to parathyroid hyperplasia, adenoma or carcinoma (table 3).38 ,39 ,41–44 ,46 ,99 ,123 ,131 With the exception of rare double adenomas, parathyroid adenomas and carcinomas are almost always uniglandular (solitary) lesions, whereas hyperplasia represents multiglandular proliferation, which can be asymmetrical and asynchronous.38 ,39 ,41–44 ,46 ,99 ,123 ,131 ,132 The presence of an atrophic rim of hypocellular non-tumorous parathyroid tissue adjacent to a cellular proliferation often allows an accurate diagnosis of ‘parathyroid adenoma’ in the appropriate clinical and biochemical setting (figure 5).38 ,39 ,41–44 ,46 ,99 ,123 ,131 However, in the absence of an atrophic rim, hyperplasia cannot be excluded at the morphologic level.38 ,39 ,41–44 ,46 ,99 ,123 ,131 For this reason, an enlarged parathyroid gland, lacking a normal rim, should be classified as an ‘enlarged cellular parathyroid gland’. If biochemical cure is achieved after surgery, a diagnosis of parathyroid adenoma can be rendered, after excluding parathyroid carcinoma. It should be noted that hyperplastic glands can also present small areas of normocellular or slightly cellular tissue indistinguishable from those surround parathyroid adenomas.38 ,39 ,41–44 ,46 ,99 ,123 ,131 Therefore, correlation with intraoperative PTH levels and postoperative PTH and calcium levels is crucial to render a definitive diagnosis of adenoma.38–46 ,99 ,123 ,131

Parathyroid adenoma. With the exception of rare double adenomas, parathyroid adenomas are almost always uniglandular (solitary) lesions. The presence of an atrophic rim of hypocellular non-tumorous parathyroid tissue adjacent to a cellular proliferation often allows an accurate diagnosis of ‘parathyroid adenoma’ in the appropriate clinical and biochemical setting.
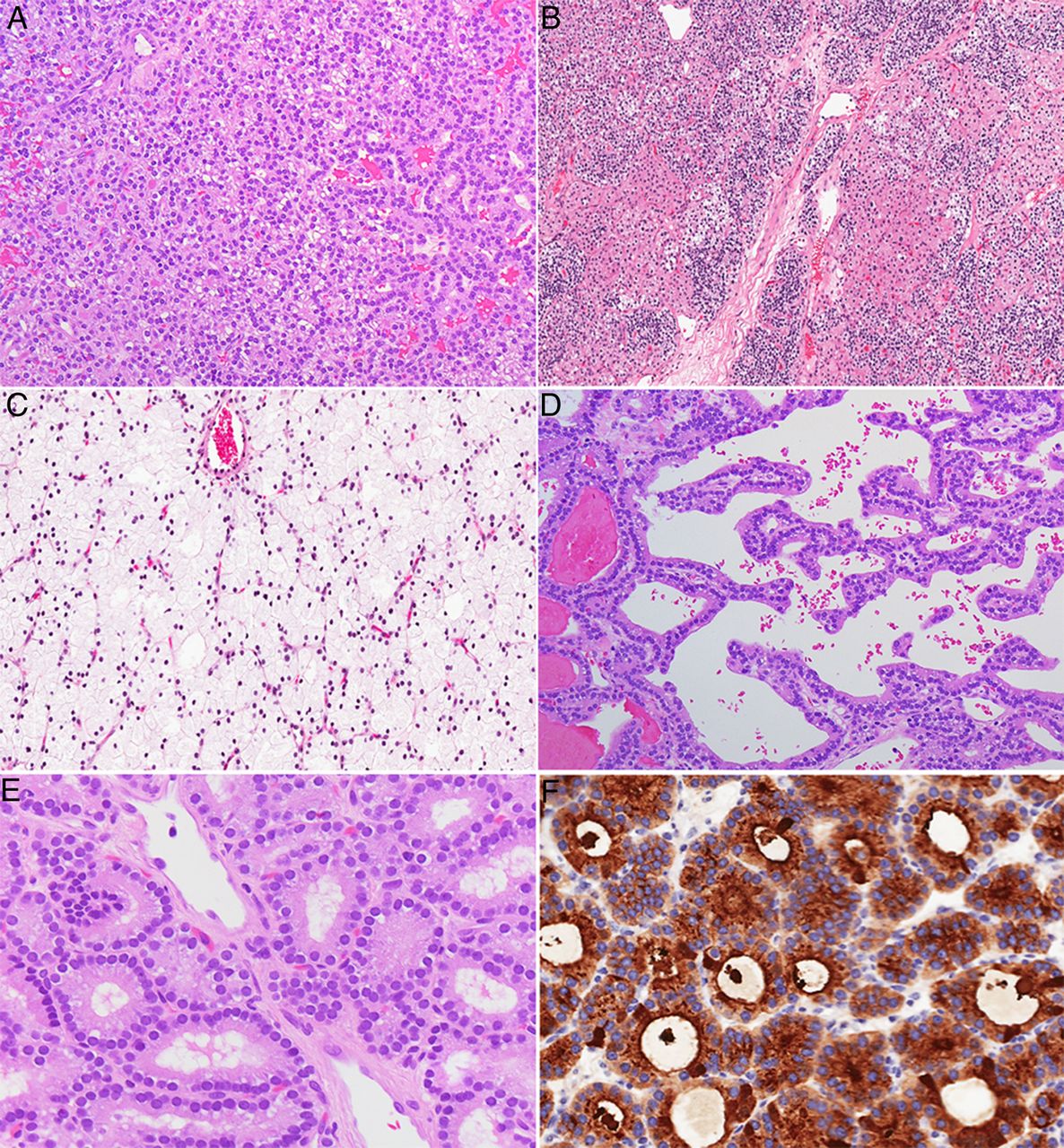
Parathyroid adenomas are by far the most common cause of primary HPT, accounting for 80–85% of cases.4 ,38 ,42 ,46 Foci of cystic change can occur in larger adenomas, particularly in the setting of a familial syndrome (ie, HPT-JT syndrome).43 ,44 ,123 Fibrosis is also seen in some patients with MEN-1 and MEN-4 syndromes, as well as following procedural interventions of the parathyroid gland(s) (eg, fine needle aspiration for PTH measurements and/or cytological examination; ethanol injection) (figure 6).38–40 ,42 ,44 ,45 ,99 ,133 Microscopically, a parathyroid adenoma is composed of varying proportions of chief, clear, transitional oncocytic and oncocytic cells.38–40 ,42 ,44 ,45 ,99 ,133 While ‘chief cell’ adenomas are the most prevalent type, rare morphological variants have also been reported, including oncocytic cell adenomas, water-clear cell adenomas and lipoadenomas.38–40 ,42 ,44 ,45 ,99 ,133 Rare cases with spindle cell features have also been described.42 Various architectural patterns have been reported, including arrangements in cords, nests, sheets, papillary, pseudopapillary and follicles with palisaded appearance around blood vessels (figure 6).38–40 ,42 ,44 ,45 ,99 ,133 Some parathyroid adenomas show prominent follicular architecture, mimicking thyroid tissue (figure 6).38–40 ,42 ,44–46 ,99 ,133 However, a distinction can be made based on the observation that the follicles in parathyroid adenomas typically do not contain birefringent calcium oxalate crystals, which are usually seen in thyroid follicles.42 ,99 ,134 Cytologically, the nuclei of tumour cells often appear round and larger than those of adjacent non-neoplastic cells.38–40 ,42 ,44–46 ,99 ,133 Occasionally, the use of immunohistochemistry (positivity for PTH, GATA3 or GCM-2) is helpful to confirm the primary parathyroid origin of an unusual neoplasm (figure 6).94 Although scattered mitotic figures can be present, the proliferative index of most adenomas, as assessed by Ki-67 (MIB-1), is generally <5%.38–40 ,42 ,44–46 ,98 ,99 ,133 ,135

Various cytological and architectural patterns associated with parathyroid adenomas. Most adenomas consist of chief cells (A), rare morphological variants have also been reported, including oncocytic cell adenomas, mixed cellular adenomas (B), water-clear cell adenomas (C), and lipoadenoma. Various architectural patterns have been reported, including arrangements in cords, nests, sheets, papillary/pseudopapillary (D) and follicles (E). Some parathyroid adenomas show prominent follicular architecture and proteinaceous material in the lumina, mimicking thyroid tissue (E). However, parathyroid proliferations with follicular growth typically lack birefringent calcium oxalate crystals, which are often seen in thyroid follicles. Occasionally, the use of immunohistochemistry (positivity for parathyroid hormone (PTH), GATA3 or GCM-2) is helpful to confirm the primary parathyroid origin in challenging cases (F; PTH immunostain).
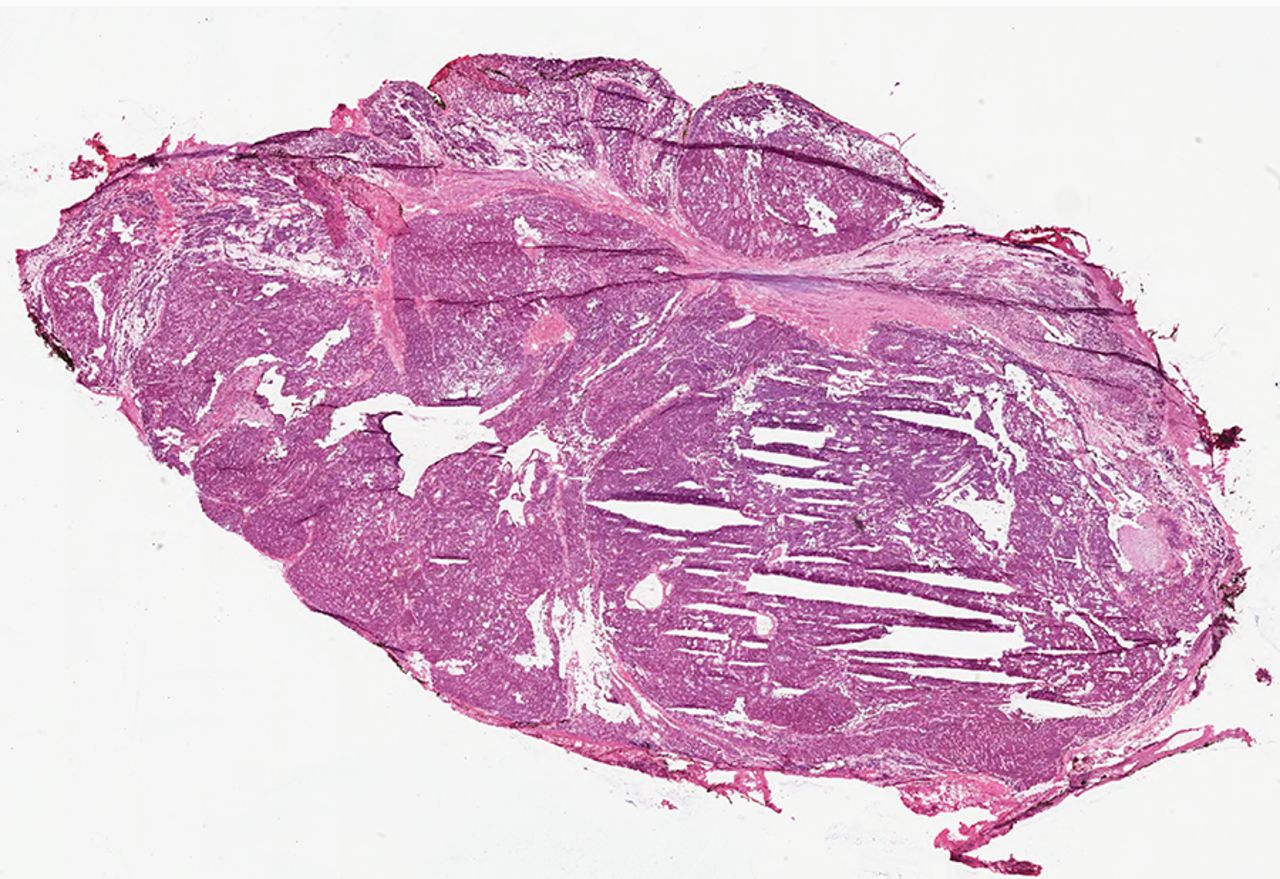
Parathyroid hyperplasia is a multiglandular disorder, causing 10–15% of cases of primary HPT.4 ,38 ,42 ,44 ,46 Three morphological variants can be seen: diffuse (diffuse hyperplasia), localised with one or more nodules (nodular hyperplasia) or a mixture of both patterns (mixed diffuse/nodular hyperplasia) (figure 7).38 ,42 ,44 ,45 ,123 The individual hyperplastic glands are grossly increased in size and weight; microscopically, they show chief, oncocytic and/or clear cells.38–40 ,42 ,44 ,45 ,99 In most cases, chief cells tend to predominate, hence the classic term ‘chief cell hyperplasia’. In rare instances, abundant stromal fat is observed, resulting in a morphological variant termed ‘lipohyperplasia’.38–40 ,42 ,44 ,45 ,99 Foci of cystic change with fibrosis and hemosiderin deposition can occur, particularly in long-standing prominent hyperplasia related to chronic renal failure.38–40 ,42 ,44 ,45 ,99 A rare ‘water-clear cell’ variant of parathyroid hyperplasia has also been reported, presenting with more florid HPT and hypercalcaemia than the common chief cell hyperplasia, often with grossly, enlarged, cystic and haemorrhagic glands.38–40 ,42 ,44 ,45 ,99 Similarly, water-cell adenomas have also been described.38–40 ,42 ,44 ,45 ,99 ,136–139 The glycogen-free water-clear cell morphology is attributed to cytoplasmic clearing due to the presence of numerous small vacuoles derived from the Golgi apparatus.38–40 ,42 ,44 ,45 ,99 ,136–139 The molecular basis underlying this ‘water-clear cell’ variant morphology remains unclear at this time.38–40 ,42 ,44 ,45 ,99 ,136–139

Parathyroid hyperplasia. Unlike parathyroid adenoma and carcinoma, parathyroid hyperplasia is a multiglandular parathyroid disease. In this photomicrograph, parathyroid hyperplasia is illustrated in a three and half parathyroidectomy specimen.
Rare cases of ‘double adenomas’ have also been described to cause multigland disease in primary HPT.42 ,44 ,46 ,123 ,132 ,140 Since the morphological distinction between adenomas and asymmetrical nodular hyperplasia is somewhat arbitrary at times, there is controversy as to whether double adenomas are truly a distinct clinical and biological entity from nodular hyperplasia.39 ,40 ,42 ,44 ,46 ,123 ,132 ,140 Furthermore, emerging molecular data suggest that the nodules in parathyroid nodular hyperplasia represent clonal proliferations that are more closely related to adenomas, making the distinction between multiple adenomas and nodular hyperplasia even more ambiguous.5 ,11 ,29 ,31 ,33 ,39 ,42 ,44 ,46 ,74 ,80 ,99 ,141–145 In fact, this is best demonstrated in patients with MEN syndromes (MEN-1, MEN-4), as well as those with tertiary HPT, who present with multiglandular adenomas; these tumours represent distinct clonal proliferations on molecular studies but are clinically thought to be hyperplasia due to multiglandular involvement.5 ,11 ,29 ,31 ,33 ,39 ,42 ,44 ,46 ,74 ,80 ,99 ,141–146 In light of these findings, the term ‘nodular parathyroid disease’, rather than nodular hyperplasia, may seem more appropriate to reflect the molecular basis of these clonal proliferations arising in a background of polyclonal diffuse hyperplasia.5 ,11 ,29 ,31 ,33 ,39 ,42 ,44 ,46 ,74 ,80 ,99 ,141–146 In other terms, this terminology may seem more accurate when designating neoplastic proliferations involving multiple parathyroid glands.
Secondary HPT is generally caused by diffuse parathyroid hyperplasia in response to prolonged reduced levels of extracellular calcium from various secondary aetiologies.7 ,11 ,42 ,44 In late stages, the emergence of an autonomous PTH-producing neoplasm (usually adenomas or rare carcinoma) from a background of secondary hyperplasia has been described, causing ‘tertiary hyperparathyroidism’.7 ,11 ,42 ,44 ,74 ,99 ,143 ,147 ,148 Clinically, this histopathological progression from diffuse to nodular parathyroid hyperplasia is associated with the development of refractory HPT and new hypercalcaemia, in patients with previously controlled secondary HPT.7 ,11 ,42 ,44 ,74 ,99 ,143 ,147 ,148 At the molecular level, this progression sequence has been linked to clonal transformation from diffuse polyclonal hyperplasia due to decreased calcium-sensing receptor (CaSR) signalling from hypocalcaemia.7 ,11 ,42 ,44 ,74 ,99 ,148–152 Morphologically, parathyroid hyperplasia with prominent nodular configuration may show areas of haemorrhage, cyst formation, fibrosis and chronic inflammation, which often correlate with the degree of HPT.7 ,11 ,42 ,44 Cytologically, chief cells tend to predominate in early phases of secondary HPT, with gradual replacement by oncocytic cells described in late stages of the disease.7 ,11 ,42 ,44
While most parathyroid tumours are benign neoplasms (adenomas), parathyroid carcinomas can occur in <1–5% of patients with HPT.4 ,7 ,39 ,40 ,42–44 ,46 ,51 ,97–99 ,102 ,123 ,153 Grossly, these tend to be larger (mean diameter of 3.4 cm and weight of 19.15 g) and can be densely adherent to thyroid or surrounding soft tissues intraoperatively.39 ,40 ,42 ,43 ,44 ,46 ,51 ,96–98 ,123 The diagnosis of malignancy is rendered when a parathyroid neoplasm exhibits any of the followings: vascular invasion (tumour cells invading the vessel wall and intravascular tumour cells admixed with thrombus), perineural invasion, invasive growth into adjacent structures/organs and/or metastasis (lymph nodes or distant organs; often lung, liver, bone) (figure 8).39 ,40 ,42–44 ,46 ,51 ,96–98 ,123 ,153 Nonetheless, it should be noted that the diagnosis of malignancy should not be based alone on the identification of the following morphological features: as broad fibrous bands, increased mitoses, nuclear atypia, necrosis and pleomorphism. These findings may also be present in some benign parathyroid disease (adenoma or hyperplasia).39 ,40 ,42–44 ,46 ,51 ,96–98 ,123 Architecturally, parathyroid carcinomas tend to show solid growth pattern with tumour cells arranged in cohesive cellular masses, although nesting or trabecular growth patterns can also occur.42–44 ,46 ,123 Their cytological composition is similar to parathyroid adenomas, with varying proportions of chief cells, oncocytes, transitional oncocytes and scattered water-clear cells.42–44 ,46 ,123 Most parathyroid carcinomas show mild-to-moderate nuclear atypia, which is indistinguishable from adenomas.39 ,40 ,42–44 ,46 ,51 ,97 ,98 ,123 Overall, the distinction between parathyroid carcinomas and adenomas is extremely difficult on cytology, with the exception of a subset of carcinomas that exhibit significant nuclear pleomorphism with macronucleoli.39 ,40 ,42–44 ,46 ,51 ,97 ,98 ,123 A triad of high-risk histopathological features (necrosis, macronucleoli, >5 mitoses per 50 high-power fields) has been reported to predict malignant behaviour in some parathyroid tumours.42 ,120 ,154

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Parathyroid carcinoma. Common morphological findings encountered in parathyroid carcinomas include the presence of broad fibrous bands (A), necrosis and solid/sheet-like growth pattern (B). In some cases, increased mitotic activity, atypical mitoses and nuclear atypia may be identified. The identification of any of these features is not diagnostic of parathyroid carcinoma. The diagnosis of malignancy is typically rendered when a parathyroid tumour shows any of the followings: (1) vascular invasion (C), (2) perineural invasion, (3) invasive growth into adjacent structures (D; computerised tomography showing a parathyroid carcinoma invading trachea) and (4) metastasis. In addition, the use of ancillary biomarkers is extremely valuable to support or exclude a diagnosis of malignancy. In particular, nuclear and/or nuclear loss of parafibromin expression is considered a diagnostic feature of parathyroid carcinoma. Please note that the vascular endothelial cells and few scattered mesenchymal cells are positive for parafibromin; however, there is global loss of nuclear parafibromin expression in a parathyroid carcinoma (E).
‘Atypical adenomas’ remain a controversial pathological entity in medicine. In the parathyroid glands, these represent borderline neoplasms showing some histopathological findings of parathyroid carcinomas (band forming fibrosis, increased mitotic activity, presence of tumour cell within a thickened capsule) but lacking the definite diagnostic features of malignancy (invasion into adjacent tissues, vascular invasion and/or metastases).38 ,40 ,42–44 ,46 ,98 ,123 ,153–156 The practising pathologist should be aware of the fact that fibrosis can occur in many situations including previous manipulations (post-FNA biopsy for PTH measurements or cytological examination, or ethanol injection), MEN1/MEN4 syndrome, lithium intake or long-standing chronic renal failure related-parathyroid hyperplasia, in the setting of parathyroiditis or in large degenerated adenomas.153 While the parathyroids do not have a true anatomic capsule, fibrotic bands at the periphery of the gland can mimic tumour cells within a thickened capsule. As discussed earlier, mitotic figures can be present in some benign parathyroid disease.153 In reality, the concept of ‘atypical parathyroid adenoma’ probably stems from the previous limits of the histopathological examination, whereby borderline parathyroid tumours with ‘equivocal’ histological features could not be classified with certainty based on morphology alone, given significant inter-observer variability as well as the lack of clinicopathological correlations in many practices.38 ,40 ,42–46 ,98 ,123 ,153 ,155–158 However, with recent developments in molecular pathology and ancillary tools, the classification of these borderline tumours has been greatly refined.153 When used in the appropriate clinical and pathological setting, ancillary biomarkers serve a crucial role in the distinction of malignancy in these borderline tumours, thereby enhancing the accuracy of the pathological examination.38 ,42 ,43 ,45 ,46 ,98 ,99 ,155 ,156 ,158–161
In diagnostically challenging cases, the use of ancillary biomarkers is extremely valuable to support or exclude a diagnosis of malignancy, while uncovering differences in molecular phenotypes and prognosis.38–40 ,42 ,43 ,46 ,98 ,99 Based on the currently available evidence and our own experiences, loss-of-expression of retinoblastoma protein (Rb), Bcl-2a, p27, parafibromin, mdm2 and APC, as well as increased MIB-1 (Ki67) proliferative index >5%, overexpression of p53 and positivity for galectin-3 (in the absence of multiglandular disease) favours a diagnosis of malignancy in a parathyroid neoplasm with worrisome histopathological features.31 ,33 ,38 ,42–46 ,49 ,87 ,98 ,99 ,141 ,153 ,155 ,156 ,158–164 In particular, the use of parafibromin immunostain (figure 8E) is very helpful to differentiate between parathyroid adenomas (intact nuclear and nucleolar parafibromin expression) and parathyroid carcinomas (complete loss of nuclear or nucleolar parafibromin expression).38 ,42 ,43 ,46 ,98 ,99 ,153 ,156 ,159 ,161 ,162 Recently, routine assessment of parafibromin staining has been proposed in all parathyroid carcinomas to help select patients for genetic testing and to predict prognosis: those with parafibromin-negative parathyroid carcinomas had a significantly higher risk of recurrence, a decreased 5-year survival of 59% and a decreased 10-year survival of 23%, which may warrant closer surveillance.47 ,158 ,162
Parathyromatosis is a rare cause of persistent or recurrent HPT after parathyroidectomy.39 ,42 ,44 ,157 ,165 Some authors believe that this entity represents implantation of benign parathyroid tissue that spilled during surgery, whereas others believe that it reflects local recurrence and/or parathyroid malignancy.39 ,42 ,44 ,157 ,165 Clinical information, presence or absence of invasive growth (especially angioinvasion) and ancillary tools may be helpful in making this distinction.39 ,42 ,44 ,157 ,165
Pathogenesis and molecular genetics of HPT
Normal physiology of PTH secretion
In normal physiology, PTH secretion is mediated by the CaSR signalling pathway.7 ,33 ,80 ,88 ,141 ,166–169 CaSR, encoded by CaSR gene, is a G-protein-coupled receptor found on the surface of parathyroid chief cells. In the presence of circulating calcium, CaSR is activated and recruits phospholipase C β, through Gq and G11.7 ,33 ,80 ,88 ,141 ,166–169 This leads to a complex series of reactions involving production of inositol triphosphate, release of calcium from intracellular stores, elevation of diacylglycerol concentrations, activation of protein kinase C, phosphorylation and internalisation of CaSR.7 ,33 ,80 ,141 ,168 ,169 Ultimately, activation of CaSR signalling pathway suppresses PTH secretion by two mechanisms: first, CaSR signalling activates Gαq and phospholipase A2, generating arachidonic acid metabolites, which have been shown to inhibit PTH secretion directly; second, vitamin D receptor (VDR) is overexpressed on parathyroid chief cells, thereby increasing their sensitivity to the negative feedback exerted by 1,25(OH)2-vitamin D, further suppressing PTH production.7 ,33 ,80 ,141 ,168 ,169 When hypocalcaemia occurs, CaSR signalling is downregulated, and this allows the parathyroid chief cells to release the PTH stored in their cytoplasmic granules, while concurrently increasing PTH gene transcription and protein synthesis.7 ,33 ,80 ,141 ,168 ,169 PTH binds to PTH/PTH-related peptide receptors at various sites in the body, stimulating osteoclast-mediated bone resorption, renal reabsorption of calcium and 1,25(OH)2D synthesis to increase calcium levels.7 ,33 ,80 ,141 ,168 ,169 With the rise of circulating calcium levels, a classic endocrine feedback loop occurs, whereby the CaSRs and its signalling pathway are reactivated, silencing PTH secretion.7 ,33 ,80 ,141 ,168 ,169 In HPT, various alterations inactivating CaSR signalling pathway can occur, causing excess PTH secretion.7 ,11 ,13 ,33 ,42 ,78 ,81 ,141 ,166 Furthermore, if downregulation of CaSR persists over time, studies have shown that it can lead to aberrant cell growth and proliferation (thought to be related to aberrant Wnt/β-catenin and cyclin D1 signalling), suggesting that CaSR signalling may serve a tumour suppressor function in the parathyroid glands.7 ,11 ,33 ,42 ,78 ,81 ,141 ,152 ,166 ,170–172
Hereditary HPT
Most patients with inherited parathyroid disease present with multiglandular involvement (ie, parathyroid hyperplasia), although rare solitary proliferations can also occur.30 ,38 ,42 ,44 ,46 ,99 ,143 Common hereditary syndromes associated with HPT include familial cancer predisposition syndromes (MEN-1, MEN-2A, MEN-4 and HPT-JT syndromes) and hereditary ‘hypercalcemic’ syndromes related to aberrant CaSR signalling (FHH-1, FHH-2, FHH-3, neonatal severe HPT and familial hypercalciuric hypercalcaemia).13 ,29 ,30 ,32 ,38 ,42 ,44 ,46 ,83 ,99 ,141 ,143 Rarely, inherited HPT can occur in a non-syndromic fashion, also known as ‘familial isolated hyperparathyroidism’, due to germline mutations of MEN1, CDC73/HRPT2, CaSR or cyclin-dependent kinase inhibitor (CDKI)-encoding genes (CDKN1B, CDKN1A, CDKN2B and CDKN2C).13 ,29 ,30 ,42 ,99 ,141 ,173
MEN syndromes represent a group of autosomal-dominant cancer predisposition syndromes.4 ,7 ,32 ,42 ,141 ,143 ,174 Three distinct types (MEN-1, MEN-2A, MEN-4) are associated with HPT.32 ,42 ,141 ,143 MEN-1 syndrome is caused by germline inactivating mutations in the tumour suppressor gene MEN1 (11q13), encoding ‘menin’ protein, which is thought to prevent nuclear translocation of β-catenin, thereby suppressing Wnt/β-catenin signalling, a well-known tumorigenesis pathway.32 ,42 ,141 ,143 ,175 Phenotypically, MEN-1 is characterised by the development of multiglandular parathyroid adenomas (90%), gastroenteropancreatic neuroendocrine tumours (60%) and pituitary adenomas (30%); additional tumours reported in this syndrome include adrenocortical tumours, facial angiofibromas, collagenomas, lipomas and/or other neuroendocrine tumours of various sites (thymus, lung, stomach).32 ,42 ,85 ,99 ,141 ,143 ,176 The mechanisms underlying tumorigenesis in this syndrome appear to be related to somatic inactivation of the wild-type allele, in addition to an inherited altered copy of the MEN1 gene.32 ,42 ,85 ,99 ,141 ,143 ,176
Recently, a subset of patients presenting with multiglandular parathyroid disease and what appeared to be MEN-1 phenotype but lacking MEN1 gene mutation were subsequently given the diagnosis of MEN-4 syndrome (also known as ‘MEN X syndrome’).42 ,85 ,98 ,99 ,141 ,143 ,176 These patients harboured inactivating germline mutations in the tumour suppressor gene CDKN1B (12p13.1; encoding the CDKI, p27kip1, implicated in cyclin D1 signalling).42 ,85 ,98 ,99 ,141 ,143 ,176 Phenotypically, patients with MEN-4 are almost indistinguishable from those with MEN-1 related disease.4 ,42 ,46 ,85 ,98 ,99 ,141 ,143 ,176 On pathological examination, both MEN-1 and MEN-4-related parathyroid proliferations may show regions of fibrosis and nodularity.39 ,42 ,44 ,46 ,85 ,99 ,176
MEN-2A syndrome is characterised by activating germline mutations in the RET proto-oncogene (10q11.2), predisposing to the development of HPT (20–30%), medullary thyroid carcinoma arising from a background of precursor C-cell hyperplasia (also known primary or neoplastic C-cell hyperplasia) and pheochromocytomas arising in adrenal medullary hyperplasia.4 ,32 ,42 ,44 ,99 ,141 ,143 ,177 ,178
HPT-JT is another autosomal-dominant cancer predisposition syndrome associated with HPT due to germline inactivating mutations of CDC73 (formerly known as HRPT2; 1q31.2).4 ,7 ,32 ,42 ,51 ,141 ,158 ,161 ,179 CDC73/HRPT2 encodes the parafibromin protein, which serves a key tumour suppressor role in the parathyroid glands, by interacting with polymerase associated factor 1 (PAF1) in histone ubiquitination/methylation, mediating gene transcription, inducing apoptosis, inhibiting cyclin D1 signalling, regulating Wnt/β-catenin signalling and growth factor gene transcription.4 ,7 ,32 ,42 ,51 ,141 ,158 ,161 ,162 ,179 Phenotypically, HPT-JT syndrome is characterised by primary HPT, as well as fibro-osseous lesions in the mandible and maxilla, and occasional uterine and renal lesions, including cysts, hamartomas, renal cell carcinoma and Wilms tumours.4 ,7 ,32 ,141 ,158 ,161 The HPT is usually attributed to multiglandular parathyroid disease, comprising of multiple parathyroid nodules/adenomas associated with cystic change; about 10–20% of these lesions eventually progress to parathyroid carcinomas, often due to bi-allelic inactivation of CDC73/HRPT2 and complete loss of parafibromin expression.4 ,31 ,42 ,51 ,141 ,158
In addition to the previously described cancer predisposition syndromes, a group of familial ‘hypercalcemic’ syndromes (FHH-1, FHH-2, FHH-3, neonatal severe HPT, familial hypercalciuric hypercalcaemia) has been reported to cause HPT, due to aberrant inactivation of CaSR signalling in the parathyroid glands, kidneys and skeletal bones.13 ,32 ,42 ,75 ,76 ,77 ,141 These conditions are generally inherited in an autosomal-dominant manner and appear to have near-complete penetrance for the phenotypical expression of hypercalcaemia.13 ,32 ,42 ,75–77 ,141 ,167 Among these, FHH type 1 is the most classic form, characterised by germline heterozygous inactivating mutations of CaSR (3q21.1), encoding CaSRs.13 ,32 ,42 ,75–77 ,141 ,167 Clinically, affected patients are typically asymptomatic and present with inappropriate hypocalciuria in the setting of overt hypercalcaemia and normal or elevated serum PTH.4 ,7 ,13 ,30 ,32 ,42 ,75–77 ,141 ,167 At the molecular level, this phenomenon is mainly attributed to mutant CaSR in the kidney, preventing the normal physiological hypercalciuric response to hypercalcaemia.4 ,7 ,13 ,30 ,32 ,42 ,75–77 ,141 Although concurrent primary HPT due to a parathyroid adenoma can occur, the majority of patients with FHH-1 do not have detectable parathyroid disease, and therefore rarely benefit from parathyroidectomy.7 ,13 ,29 ,30 ,34 ,42 ,141 ,180 ,181 Pathologically, parathyroid enlargement (hyperplasia) has been reported in 15–20% of cases.3 ,32 ,42 ,44 ,83 ,182 ,183
In addition to FHH-1, a severe neonatal phenotype (neonatal severe primary HPT) has been described to cause multiglandular parathyroid disease due to germline homozygous inactivating mutations of the CaSR gene.4 ,7 ,13 ,29 ,42 ,141 Clinically, patients with this condition typically present shortly after birth, with life-threatening hypercalcaemia, hypocalciuria and severe HPT.4 ,7 ,13 ,29 ,30 ,42 ,141 In contrast to FHH-1, neonatal severe HPT generally requires urgent total parathyroidectomy to prevent a fatal outcome in affected patients.5 ,34 ,34 ,36 On pathological evaluation, the parathyroid glands are visibly enlarged, hyperplastic and hypercellular.4 ,7 ,13 ,30 ,32 ,42 ,44 ,46
A milder form, known as familial hypercalciuric hypercalcaemia (or autosomal-dominant moderate HPT), has also been reported to cause HPT due to germline inactivating mutation in the intracytoplasmic tail domain of the CaSR gene.13 ,30 ,42 ,141 ,184 Clinically, affected patients present with elevated calcium and PTH levels, an appropriate hypercalciuric response, hypermagnesemia and hyperphosphaturia.13 ,30 ,42 ,141 ,184 Normalisation of calcium levels has been described in some cases after subtotal parathyroidectomy.13 ,30 ,42 ,44 ,141 ,184 Pathologically, multiglandular parathyroid disease (ie, nodular hyperplasia) is observed in most cases.13 ,30 ,42 ,44 ,141 ,184
Recently, two novel forms of FHH (types 2 and 3) have been uncovered.13 ,32 ,88 ,90 ,91 ,141 ,185 FHH-2 and FHH-3 are associated, respectively, with germline inactivating mutations of GNA11 (19p13.3) and AP2S1 (19q13.2) genes.13 ,32 ,88 ,90 ,91 ,141 ,185 GNA11 gene encodes the α-subunit of G11, one of the principal G proteins activating CaSR signalling pathway, whereas AP2S1 gene encodes the adaptor protein 2 σ-subunit involved in CaSR endocytosis.13 ,32 ,88 ,90 ,91 ,141 ,185 Both mutations in GNA11 and AP2S1 genes cause hypocalciuric hypercalcaemia though aberrant inactivation of CaSR signalling, similar to FHH-1.13 ,32 ,88 ,90 ,91 ,141 ,185 Phenotypically and pathologically, patients with FHH-2 typically present with features indistinguishable from FHH-1, whereas those with FHH-3 tend to present with osteomalacia, higher levels of PTH and more frequent parathyroid gland involvement (hyperplasia).3 ,13 ,32 ,42 ,88 ,90 ,91 ,141 ,185
Sporadic parathyroid adenomas
Most parathyroid adenomas are sporadic neoplasms.39 ,42–44 ,141 Although the majority of cases do not have an identifiable risk factor, epidemiological studies suggest that a previous history of ionising radiation predisposes to the formation of sporadic parathyroid tumours.4 ,7 ,38 ,42 Somatic alterations in MEN1 and CCND1/PRAD1 genes have emerged as important drivers in the development of 25–40% of sporadic parathyroid adenomas.42 ,46 ,81 ,141 ,186 ,187
As discussed previously, MEN1 and its gene product, menin, are thought to serve a tumour suppressor role by regulating Wnt/β-catenin signalling, a well-documented pathway in tumorigenesis.141 ,175 ,188 Although an inherited mutant copy of the MEN1 gene may be sufficient to cause multiglandular parathyroid disease (hyperplasia) and HPT (in MEN-1 syndrome or isolated familial HPT), an additional somatic inactivating mutation in the remaining copy of MEN1 (ie, the wild-type allele) is generally required for the development of sporadic parathyroid tumours (>25% in some series).42 ,81 ,141 Alternatively, pure somatic mutations can result in bi-allelic inactivation of MEN1, seen in some parathyroid adenomas.141 ,189 ,190 These findings are consistent with comparative genomic hybridisation reports, where loss of 11q (locus of MEN1) is the most frequent alteration in adenomas; losses of 1p, 6q, 9p, 11p, 13q and 15q (possible loci for undiscovered tumour suppressor genes) and gains in 7, 16p and 19p (possible loci for undiscovered proto-oncogenes) have also been reported.42 ,141 ,191
CCND1/PRAD1 (11q13) is a proto-oncogene, encoding cyclin D1 protein, a holoenzyme thought to inactivate the tumour-suppressor retinoblastoma protein (Rb), inducing cellular proliferation.31 ,33 ,80 ,81 ,141 ,192–196 Somatic activating alterations of CCND1/PRAD1 gene, causing excess cyclin D1 signalling, have been reported in 20–40% of sporadic parathyroid adenomas.31 ,33 ,80 ,81 ,141 ,192–196 Deregulation of CCND1/PRAD1 gene expression can occur as a result of rearrangements with PTH promoter gene (∼8%) and/or other enhancers/promoters, leading to amplification or transcriptional activation of cyclin D1.31 ,33 ,38 ,46 ,80 ,81 ,141 ,192–196
Concurrent to the discovery of germline mutations in CDKI-encoding genes causing familial HPT (in MEN-4 syndrome and isolated familial HPT), somatic alterations of CDKI-encoding genes (CDKN1B, CDKN1A, CDKN2B, CDKN2C) were also identified in sporadic parathyroid adenomas.31 ,33 ,38 ,46 ,80 ,81 ,141 ,173 ,197 Of these, somatic inactivating mutations of the CDKN1B gene (encoding p27kip1) causing excess cyclin D1 signalling have been well described, and this finding was further corroborated by studies showing decreased expression of p27 in sporadic parathyroid adenomas at both the RNA and protein levels, supporting its role in parathyroid tumorigenesis.42 ,141 ,197–199
Despite its well-documented role in hereditary HPT, somatic mutations of the CaSR gene have not been identified in sporadic disease.13 ,42 ,76 ,141 ,152 ,200 Nonetheless, several studies reported that a significant subset of sporadic parathyroid adenomas display aberrant CaSR signalling, characterised by reduced expression of VDRs and CaSRs.13 ,42 ,76 ,141 ,150 ,152 ,172 ,200–204 The exact mechanism underlying loss of expression of VDR and CaSR remains unclear, although genetic and epigenetic alterations in their respective genes have been sought but not found.13 ,42 ,76 ,141 ,150–152 ,172 ,200–204 Given the fact that the pathogenesis of >50% of sporadic parathyroid adenomas remains unknown, these findings certainly raise the possibility that undiscovered somatic alterations, causing deregulation of CaSR signalling (either upstream or downstream of CaSR and VDR), may play an important role in parathyroid tumorigenesis.13 ,42 ,76 ,141 ,150 ,152 ,172 ,200–205
The recent use of whole-exome sequencing in parathyroid adenomas revealed additional somatic mutations, involving ZFX (∼5%; a putative proto-oncogene thought to be a downstream target of cyclin D1); CTNNB1 (2–5%; encoding β-catenin), EZH2 (∼1%; a putative oncogene involved in histone methyltransferase activity and thought to cause aberrant accumulation of β-catenin) and POT1 (<1%; regulator of telomere integrity and genome stability).82 ,141 ,186 ,187 ,206–209
Over the past few years, DNA methylation and microRNA profiling studies have uncovered important epigenetic alterations in sporadic parathyroid tumours.141 ,152 ,205 ,210–212 When compared to normal parathyroid tissue, parathyroid neoplasms were shown to have aberrant hypermethylation of tumour suppressor genes in Wnt/β-catenin signalling (APC, SFRP1, SFRP2, SFRP4), cyclin D1 signalling (CDKN2A, CDKN2B), as well as WT1, RIZ1, RASSF1A and HIC1.141 ,152 ,205 ,210–213 As mentioned previously, cyclin D1 signalling plays an important role in cell cycle progression in parathyroid tissue and can be induced by Wnt/β-catenin signalling, another tumorigenesis pathway in many solid tumours.81 ,141 ,196 ,212 ,214–216 APC, SFRP1, SFRP2 and SFRP4 genes all serve important regulatory functions in Wnt/β-catenin signalling, whereas CDKN2A and CDKN2B genes appear to regulate cyclin D1 signalling.81 ,141 ,212 ,214–216 WT1 (Wilms tumour 1) encodes an important transcription factor for cellular development and survival, and mutations in WT1 have been linked to various solid tumours.81 ,141 ,210 ,212 Similarly, RIZ1 (retinoblastoma-interacting zinc finger gene) appears to be involved in cell cycle regulation.141 ,205 ,210 ,212 ,217 RASSF1A (Ras association domain family 1 isoform A; 3p21) is a component in the Ras signalling pathway and has been shown to suppress cyclin D1.218–220 HIC1 (17p13) is a putative tumour suppressor gene, epigenetically deregulated in parathyroid tumours due to histone H3 lysine modification.212 ,213 Recently, aberrant expression of a panel of embryonic-related microRNAs has been described in a series of sporadic parathyroid tumours, suggesting a role for microRNA deregulation in parathyroid tumorigenesis.211 ,212
Sporadic parathyroid carcinomas
In contrast to parathyroid adenomas, parathyroid carcinomas are aggressive sporadic neoplasms, with significant morbidity and mortality related to PTH-mediated hypercalcaemia.7 ,19 ,42 ,47 ,51 ,97 Consequently, the study and understanding of their molecular biology is important to uncover novel diagnostic, prognostic and predictive biomarkers to improve patient care. With recent advances in molecular pathology, a wide array of genetic and epigenetic alterations, as well as biomarkers, have been uncovered in parathyroid cancer.42 ,98 ,141 ,156 ,158 ,163 ,210 ,216
Inactivation of CDC73/HRPT2 (1q31.2) and its gene product, parafibromin, is a major driver in parathyroid cancer, appearing in >70% of cases in some series.42 ,98 ,141 ,156 ,158 ,161 As mentioned previously, parafibromin serves critical tumour suppressor functions in normal parathyroid tissue, notably by interaction with PAF1 complex and regulating cyclin D1 and Wnt/β-catenin signalling.31 ,42 ,98 ,141 ,156 ,158 ,161 ,221 It should be noted that decreased expression of parafibromin can occur in rare parathyroid adenomas (often with cystic features; associated with germline CDC73/HRPT2 mutations).42 ,46 ,98 ,141 ,156 ,158 ,161 ,221 However, complete loss of parafibromin expression, due to bi-allelic inactivating mutations of CDC73/HRPT2, is almost pathognomonic for parathyroid carcinomas as it is almost never seen in adenomas.42 ,46 ,98 ,141 ,156 ,158 ,161 ,162 ,221–224 Furthermore, all patients with histologically proven parathyroid carcinomas with loss of parafibromin staining should be considered for genetic testing since up to 20% of cases may harbour germline mutations in CDC73/HRPT2 with or without clinical features of HPT-JT syndrome.39 ,42–46 ,98 ,141 ,156 ,158 ,161 ,162 ,221–224
Additional molecular alterations identified in parathyroid carcinomas (CCND1, CDKIs, MEN1, EZH2, RIZ1, RASSF1, GSK3B, WT1, APC, SFRP, HIC1, CaSR, VDR and microRNA deregulation) are non-specific and can also occur in parathyroid adenomas.31 ,42 ,43 ,81 ,82 ,141 ,151 ,164 ,205 ,210 ,211 ,212 ,216 ,217 ,219 Of note, MEN1 mutations, one of the key drivers in parathyroid adenomas (20–40%), appear to be less frequent in carcinomas (10–15%).31 ,42 ,81 ,141 ,225–229 Recently, whole-genome sequencing uncovered additional mutations in parathyroid carcinomas, implicating PRUNE2 (∼18%), mTOR, MLL2, THRAP3 and PIK3CA genes.141 ,226 ,227 Overexpression of PGP9.5 (gene product of UCHL1 gene), galectin-3 and TERT (telomerase reverse transcriptase gene) were also described in some parathyroid carcinomas.160 ,223 ,230 Previous comparative genomic hybridisation and molecular allelotyping studies on parathyroid carcinomas also revealed losses on chromosomes 1p, 3, 13q and 14 and gains on chromosomes 1q and 16.42 ,141 Interestingly, some of these loci (1p, 13q, 16) were also reported in adenomas.141 ,191 Several studies suggest a possible involvement of the p53/Rb signalling pathway: while TP53 mutations are rare, allelic loss and abnormal expression of p53 and retinoblastoma protein have been uncovered in a subset of parathyroid carcinomas.81 ,98 ,141
Treatment and prognosis
The management of HPT depends largely on its aetiology.1 ,4 ,6 ,7 ,29 ,34 ,35 ,36 ,50 ,107 ,231 After excluding reversible secondary aetiologies, first-line treatment of irreversible secondary HPT (often related to chronic kidney disease) mainly involves medical therapies, using low-phosphate diet, phosphate binders, 1,25-dihydroxyvitamin D3 (calcitriol or analogues; to suppress PTH secretion) and/or calcimimetic agent (cinacalcet; an allosteric activator of CaSR).1 ,4 ,6 ,7 ,11 ,29 ,34–36 ,50 ,107 ,231 ,232 In severe cases of secondary HPT refractory to medical treatment or in tertiary HPT, parathyroidectomy can be beneficial for both biochemical abnormalities and symptomatic control.1 ,4 ,6 ,7 ,11 ,29 ,34–36 ,50 ,72 ,74 ,107 ,231–234
Although percutaneous ethanol ablation may be an alternative to parathyroidectomy in select cases of primary HPT,235 ,236 surgical excision of abnormal ‘hyperfunctioning’ parathyroid gland(s) remains the only curative treatment.1 ,4 ,7 ,29 ,34–36 ,231 While symptomatic cases are addressed with parathyroidectomy, surgery in asymptomatic HPT is typically reserved to patients with one of the following conditions: age (<50 years), kidney disease (creatinine clearance <60 mL/min; radiographic nephrolithiasis/nephrocalcinosis; 24-h urinary calcium >400 mg/d with increased stone risk), bone disease (osteoporosis, ie, bone mineral density T score <−2.5 at distal 1/3 of radius, femoral neck, total hip, lumbar spine; radiographic vertebral fractures), overt hypercalcaemia (serum calcium >1.0 mg/dL above upper limit of normal) and if routine surveillance is not feasible.1 ,4 ,7 ,29 ,34–36 ,231
In all cases of primary HPT, an underlying genetic syndrome should be considered, especially if hypocalciuria or multiglandular parathyroid disease is detected.4 ,7 ,13 ,29 ,30 ,42 In particular, familial hypocalciuric hypercalcaemia should be excluded, using urinary calcium-to-creatinine clearance ratio and/or genetic testing (CASR, GNA11, AP2S1), given the fact that most patients with this condition do not benefit from parathyroidectomy.4 ,7 ,13 ,29 ,30 ,32 ,42 Furthermore, patients with MEN-1, MEN-2A or familial isolated HPT merit closer monitoring, given their increased risk of recurrent/persistent disease due to multigland parathyroid involvement.4 ,7 ,13 ,29 ,30 ,42 Sporadic primary HPT is usually caused by single-gland parathyroid disease, commonly from a solitary adenoma.4 ,7 ,13 ,29 ,30 ,38 ,42 ,123 Minimally invasive parathyroidectomy is an increasingly popular approach, although open surgery with bilateral cervical exploration is still advocated by some endocrine surgeons, particularly in multigland disease and/or recurrent HPT.4 ,7 ,13 ,29 ,30 ,42 ,123 The complete removal of abnormal parathyroid tissue should be confirmed biochemically with intraoperative (or postoperative) PTH measurements.4 ,13 ,29 ,30 ,42 ,123 Following surgery, most patients with clinically and pathologically confirmed parathyroid adenoma are cured.4 ,6 ,7 ,13 ,29 ,30 ,42 ,123
In parathyroid carcinomas, an oncological surgical approach, including at least en bloc resection (either as a primary or revision surgery), is paramount for disease control.29 ,36 ,47 ,51 ,94 ,97 Adjuvant radiation has been proposed to decrease local recurrence rate, although this approach remains controversial.47 ,51 ,94 ,97 Given its rarity, challenging diagnosis and high-risk of recurrence, suspected cases of parathyroid carcinomas should be referred to endocrinologists, surgeons and pathologists with extensive experience in endocrine oncology.29 ,36 ,47 ,51 ,94 ,97 At our institution, in a cohort of 16 patients with parathyroid cancer, the 5-year and 10-year disease-specific survival rates were 100% and 80%, respectively; the 5-year and 10-year disease-free survival rates were 69% and 43%.97 These findings confirm the relatively indolent behaviour of parathyroid malignancies reported in the literature, while reinforcing the high risk of disease recurrence, which can occur many years after initial treatment.47 ,51 ,94
In patients diagnosed with a parathyroid carcinoma, the tumour should be further tested for loss of parafibromin expression.42 ,47 ,158 ,159 ,161 ,162 Those showing loss of immunohistochemical parafibromin expression should be offered genetic testing for germline CDC73/HRPT2 mutations.42 ,47 ,158 ,162 In patients with primary HPT who cannot or do not want to undergo surgery, ethanol injection and alternative treatment modalities remain limited at this time.47 ,51 Emerging medical therapies, similar to those used in secondary HPT, have been shown to be beneficial, although not curative, in a subset of patients.1 ,47 ,231 ,232 ,237–241
Conclusion
HPT is a common, morbid yet fascinating disorder in the evolving field of endocrinology. Clinicopathological correlates of primary HPT include parathyroid adenoma (80–85%), hyperplasia (10–15%) and carcinoma (<1–5%). The clinical and biochemical history, including intraoperative PTH measurements, number, size and weight of the affected gland(s), are crucial parameters to consider when rendering a clinicopathological diagnosis of parathyroid proliferations. Ultimately, the role of the pathologist pertains to the accurate distinction between single-gland (usually a solitary parathyroid adenoma) and multigland disease (diffuse and/or nodular parathyroid hyperplasia), all the while excluding a primary parathyroid malignancy. New developments in diagnostic imaging and intraoperative rapid PTH assays have substantially improved our ability to distinguish between single-gland and multigland parathyroid disease preoperatively. These findings have tremendous implications on the prognosis of the patient, as well as the need to pursue further genetic testing, clinical monitoring and/or adjuvant therapies.242 In addition, important mechanisms underlying the molecular biology of HPT have been elucidated with the discovery of genetic and epigenetic alterations implicating cyclin D1 signalling (CCND1/PRAD1, CDC73/HRPT2, CDKN1B, CDKIs, ZFX), Wnt/β-catenin signalling (MEN1, APC, EZH2, CTNNB1, SFRPs) and CaSR signalling (CaSR, GNA11, AP2S1), offering promising targets for the development of new diagnostic, prognostic and predictive biomarkers.
Take home messages
Pathological correlations of hyperparathyroidism include parathyroid hyperplasia, adenoma and carcinoma.
The primary role of the intraoperative pathological consultation is to confirm the presence of parathyroid tissue in the resected specimen and to confirm the presence of abnormal (enlarged cellular) gland.
Clinical and biochemical features, including intraoperative PTH levels, number, weight and size of the affected parathyroid gland(s), are crucial parameters to consider when rendering an accurate diagnosis of parathyroid proliferations.
The diagnosis of parathyroid carcinoma is rendered when a parathyroid neoplasm exhibits any of the followings: vascular invasion, perineural invasion, invasive growth into adjacent structures/organs, and/or metastasis.
The application of ancillary tools can also help to distinguish benign disease from parathyroid carcinoma. In particular, the use of parafibromin immunostain is very helpful to differentiate between parathyroid adenomas (intact nuclear and nucleolar parafibromin expression) and parathyroid carcinomas (complete loss of nuclear or nucleolar parafibromin expression.
Genetic and epigenetic alterations resulting in deregulation of cyclin D1, Wnt/β-catenin and CaSR signalling have been uncovered in a significant subset of patients with primary hyperparathyroidism.
In the absence of secondary causes of hyperparathyroidism, an underlying familial syndrome should be considered in patients with parathyroid hyperplasia and carcinoma, which may be found in respectively ∼25% and ∼15% of cases.
References
Footnotes
Handling editor Runjan Chetty
Contributors OM: substantial contributions to the conception or design of the work; or the acquisition, analysis or interpretation of data for the work; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. KD, KGH and OM: drafting the work or revising it critically for important intellectual content.
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.