Article Text

Abstract
Although vasculitis involving the gastrointestinal tract (GIT) is an uncommon occurrence, occasionally vasculitis can present as haemorrhagic infarction or ischaemia for which a length of bowel is removed. Invariably, the appropriate clinical history is not forthcoming, or vasculitis is not clinically suspected. The purpose of this overview is to provide the practising gastrointestinal (GI) pathologist with a framework to recognise and diagnose vasculitides within the GIT. The classification may be approached by aetiological agent or size of vessel involved; an international consensus group now favours the latter approach. The symptoms that systemic and/or localised vasculitis may cause in the GIT are protean and non-specific. As a result, pathologists examining resection specimens for unexplained haemorrhagic infarction or ischaemia should be aware that vasculitis may be a potential cause. Several well-known systemic vasculitides such as polyarteritis nodosa, microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis or Churg-Strauss syndrome and granulomatosis with polyangiitis or Wegener's granulomatosis can occur in the GIT. The latter three constitute the antineutrophil cytoplasmic antibody-positive vasculitides. In addition, the so-called solitary organ vasculitis (SOV) can occur in the GIT as the harbinger of later onset systemic vasculitis, and be the cause of the GIT symptoms. In addition, SOV can occur incidentally and coexist with GIT disease such as gallstones or polyps, and there may be no manifestations of systemic vasculitis for years, or not at all.
- VASCULITIS
- GASTROINTESTINAL DISEASE
- DIAGNOSTICS
Statistics from Altmetric.com
Diagnosing vasculitis is usually within the practice remit of renal and/or cardiovascular pathologists, especially in patients who have known systemic vasculitis and the kidneys, for instance, are involved.
However, gastrointestinal pathologists can encounter vasculitis manifesting in the gastrointestinal tract (GIT) ab initio, and as such should be aware of the major causes and manifestations of primary vasculitis presenting in the GIT.
Thus, the purpose of this overview is to highlight the common and/or selected vasculitides that the practising gastrointestinal (GI) pathologist is likely to encounter and provide an approach to reaching a working diagnosis, if not a definitive diagnosis. Only the pertinent, salient clinicopathological features with a strong bias to the GIT will be highlighted to allow the practising GI pathologist a framework on which to build a definitive diagnosis.
Clinical information, basic stains and handling of specimens
In general, the specimen is sent for pathological evaluation without a history of vasculitis, especially if the patient does not have a clinical history of systemic vasculitis. In other words, the vasculitis of the GIT may be the sentinel lesion of systemic vasculitis, or the GIT vasculitis may be the only manifestation of vasculitis. In spite of this, the clinical history is of paramount importance in correlating the vasculitis and potential causes. The clinical history should include anything that could allude to a systemic vasculitis such as renal function, lung symptoms, serum immunoglobulin profile, tissue immune complex status (if known) and details of the patient's drug history. It is worth remembering that vasculitis is frequently a systemic, multiorgan disease with overlapping clinical and pathological features.
The cut up or grossing of the specimen ab initio should be routine and per standard protocols for ischaemic bowel disease or haemorrhagic infarcted bowel if these were the presenting features. If a clinical history of vasculitis with bowel involvement is known at the time of initial dissection, then more liberal sampling of the mesentery and its vessels should be undertaken. More often than not, in the absence of a pertinent clinical history, extra sections will be taken after initial screening of the case and a light microscopic suspicion of vasculitis on the part of the pathologist. Either way, there should be extensive sampling of more viable bowel and/or mesentery to ensure that there is unequivocal vasculitis present.
As regards stains, we feel that a good H&E stain should suffice in the vast majority of vasculitides. Some advocate the use of haematoxylin phloxine saffron stains, but it is debateable whether this has any particular advantage over the standard H&E stain. Supportive/ancillary stains to demonstrate vessel wall damage and to ascertain the type and calibre of vessel involved include elastic trichrome or elastic van Gieson or Movat pentachrome stains.
It is important to remember that vessels in the vicinity of an infarct or haemorrhagic necrosis are often necrotic and contain thrombi. This should not be taken as evidence of vasculitis. Vessels should be assessed away from areas of haemorrhage and/or infarction, ideally in the deeper layers of the bowel wall and in the mesentery.
What is vasculitis?
The quintessential definition of vasculitis is inflammation, with or without fibrinoid necrosis of the blood vessels, a process that includes arteries, veins and capillaries.
The process may involve individual vessel types selectively (ie, artery or vein or capillary/venule only) or all vessels together.
Classification of vasculitis
There are several ways vasculitis can be approached or classified: primary, secondary, according to the size of vessel involved or by the aetiological agent if known.
The GIT may be the primary site of involvement of several systemic vasculitides (see box 1).
Vasculitides that can occur primarily in the GIT
Henoch-Schonlein purpura
Polyarteritis nodosa
Churg-Strauss syndrome
Microscopic polyangiitis (rare in GIT)
Wegener's granulomatosis (rare in GIT)
Behcet's disease
Systemic lupus erythematosus
Mixed/undifferentiated connective tissue disease
Systemic sclerosis
Drugs
GIT, gastrointestinal tract
If one takes an aetiological approach, then there are three categories that may be invoked broadly (see box 2). Direct infective causes occur occasionally in the immunocompromised patient where the infective organism such as mucormycosis or aspergillosis, for instance, is angioinvasive and causes a vasculitis with subsequent GI symptoms. Most other known triggers of GI vasculitis have an immunological basis: immune complex-mediated (hepatitis B and C, systemic lupus erythematosus (SLE) rheumatoid arthritis and drug-induced). The trilogy of antineutrophil cytoplasmic antibody (ANCA)-mediated vasculitides includes Wegener's granulomatosis (WG), Churg-Strauss syndrome (CSS) and microscopic polyangiitis (MPA). Specific directed antibodies to the glomerular basement membrane result in Goodpasture's syndrome in the kidney, while antiendothelial antibodies cause Kawasaki disease, which results in a small-vessel vasculitis similar to Henoch-Schonlein purpura.
An aetiological approach to vasculitis
Direct infection
Bacterial, rickettsial, spirochetal, fungal
Immunological
Immune complex mediated
SLE and rheumatoid arthritis
Drug-induced
Henoch-Schonlein purpura
Cryoglobulinaemic vasculitis
Antineutrophil cytoplasmic antibody mediated
Wegener's granulomatosis
Churg-Strauss syndrome
Microscopic polyangiitis
Direct antibody attack mediated
Goodpasture syndrome (anti-GBM antibodies)
Kawasaki disease (antiendothelial antibodies)
Cell-mediated
Allograft organ rejection
Unknown
Giant cell (temporal) arteritis
Takayasu's arteritis
Polyarteritis nodosa
GBM, glomerular basement membrane; SLE, systemic lupus erythematosus
Boxes 1 and 2 represent two different ways of classifying vasculitis. However, the second International Chapel Hill Consensus Conference of vasculitis endorsed vasculitis being classified according to the size of vessel involved.1 This is outlined in box 3. They also suggested a nomenclatural change for CSS, now known as eosinophilic granulomatosis with polyangiitis (EGPA), and WG which is now called granulomatosis with polyangiitis (GPA).
International Chapel Hill Consensus Conference 20121
Large vessel vasculitis
Takayasu arteritis
Giant cell arteritis
Medium-vessel vasculitis
Polyarteritis nodosa (PAN)
Kawasaki disease
Small-vessel vasculitis
Antineutrophil cytoplasmic antibody-associated vasculitis (AAV)
Microscopic polyangiitis
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss)
Granulomatosis with polyangiitis (Wegener's)
Immune complex vasculitis
Antiglomerular basement membrane disease (Goodpasture)
Cryoglobulinaemic vasculitis
IgA vasculitis (Henoch-Schonlein)
Hypocomplementemic urticarial vasculitis (anti-C1q)
Variable vessel vasculitis
Behcet's vasculitis
Cogan's vasculitis
Single-organ vasculitis
Cutaneous leucocytoclastic vasculitis
Cutaneous arteritis
Primary central nervous system vasculitis
Isolated aortitis
Isolated PAN-like
Vasculitis associated with systemic disease
Lupus vasculitis
Rheumatoid vasculitis
Sarcoid vasculitis
Mixed connective tissue disorder
Vasculitis associated with probable aetiology
Hepatitis C virus-associated cryoglobulinaemic vasculitis
Hepatitis B virus-associated vasculitis
Syphilis-associated vasculitis
Drug-associated immune complex vasculitis
Drug-associated AAV
Cancer-associated vasculitis
Medium-vessel vasculitis
Polyarteritis nodosa
Polyarteritis nodosa (PAN) is mainly a medium-sized (occasionally small) vessel vasculitis that affects males in their fourth and fifth decades twice as commonly as females.2 ,3 This vasculitis generally spares veins and is ANCA-negative (see later). An autoimmune pathogenesis has been postulated for PAN; Hepatitis B and C cause a minority of cases in injection drug abusers. A number of cases are also idiopathic in aetiology.
Symptoms include fever, sweats, weight loss and muscle/joint pain. The vasculitis is distributed mainly in the vessels in the skin, kidney, nerves and GIT. Up to 50% of patients with PAN present with GI symptoms: acute abdominal pain, bleeding, haemorrhage, infarction, peritonitis, cholecystitis, appendicitis, melaena, haematochezia and perforation. This may mimic inflammatory bowel disease clinically, especially with abdominal pain after meals.
Pathology
There is transmural necrosis of the artery with fibrinoid necrosis, acute inflammation, eosinophils and lymphocytes. The brunt of the inflammation is centred on the media; all three layers of the vessel are inflamed.
Antineutrophil cytoplasmic antibodies
Before addressing some of the vasculitides, by way of introduction and to provide a basic understanding, we will mention some pertinent points regarding ANCA. ANCA are specific antibodies for antigens in cytoplasmic granules of neutrophils and monocyte lysosomes.4
This is a sensitive but not highly specific serological test for vasculitis. Nonetheless, ANCA positivity and pattern of positivity form the basis for subclassifying some vasculitides. Initially, these antibodies were detected by indirect immunofluorescence; however, immunohistochemistry has supplanted immunofluorescence, as it is more specific and clinically relevant.5 Nomenclature sometimes seen is PR3-ANCA (antiproteinase 3 ANCA) and MPO-ANCA (myeloperoxidase ANCA).
There are two patterns:
ANCA with cytoplasmic staining (c-ANCA)
This pattern is specific for immunogenic epitopes of antiproteinase-3 (PR-3) and has 98% sensitivity for GPA/WG.
ANCA with perinuclear staining (p-ANCA)
Perinuclear staining with ANCA has an antigenic specificity for epitopes expressed by MPO and is 75% sensitive for EGPA/CSS. This pattern is also found in GPA/WG, MPA and PAN. p-ANCA is important for the diagnosis of EGPA, though it is not as specific as c-ANCA is for GPA.
ANCA-positive vasculitides
There are three ANCA-positive, fibrinoid, necrotising inflammatory, leucocytoclastic, systemic, small-vessel vasculitides (see table 1). EGPA, GPA and MPA show some overlap and are sometimes clustered together under the rubric of ‘primary systemic vasculitides’.
Eosinophilic granulomatosis with polyangiitis/Churg-Strauss syndrome
Clinicopathological characteristics of the major GIT vasculitides (adapted from Ahn et al)3
This is the least common of the three ANCA-positive vasculitides, and a microbial superantigen may play a pathogenetic role. It is characterised by severe asthma, fever, eosinophilia, cardiac and renal failure, peripheral neuropathy, pulmonary infiltrates, sinusitis, purpura, subcutaneous nodules and hypertension. Specific to the GIT, abdominal pain and bloody diarrhoea are the two most common symptoms. It tends to occur in the age group of 40–60 years with a very slight predilection for males.
The second International Chapel Hill Consensus Conference recommend that EGPA be defined as an ‘eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small and medium vessels, and associated with asthma and eosinophilia. ANCA positivity is more frequent when glomerulonephritis is present’.1 Invariably, these patients have blood eosinophilia >1500/mL, or eosinophils constitute 10% of the total white blood count.
Pathology
The vasculitis of EGPA is centred on small vessels: capillaries/venules/arterioles and, very occasionally, medium-sized arteries and veins may also be involved. The typical histological appearance is of perivascular eosinophils resulting in an ‘arteriopathy’ rather than a true vasculitis. In addition, epithelioid and giant cells are found around vessels, the so-called ‘extravascular granulomas’. The true vasculitis resembles PAN with granulomatous fibrinoid necrosis of media in affected vessels. The GIT is affected in about 40% of cases of EGPA, but there is a predilection for vessels in the lung, heart, skin, central and peripheral nervous systems and kidneys. When there is mesenteric vasculitis due to EGPA, the histological findings are very similar to what is encountered in PAN.
It is preferentially a vasculitis involving small vessels: capillaries, venules and arterioles. It should be noted that medium-sized vessels might also be affected. The vasculitis is characterised by an eosinophil-rich granulomatous inflammatory response that spills over into surrounding tissue as well (figure 1A, B).

(A and B) Eosinophilic granulomatosis with polyangiitis (EGPA) or Churg-Strauss syndrome of the gastrointestinal tract with numerous eosinophils present in the inflammatory infiltrate (A). As a reflection of the hypereosinophilia associated with this vasculitis, several Charcot-Leyden crystals are noted (B) (arrow). The crystals are characteristic but not pathognomonic of EGPA.
The differential diagnosis encompasses all diseases that are characterised by eosinophilia and a vasculitis. It includes idiopathic hypereosinophilic syndrome, parasites, hypersensitivity to drugs, GPA and MPA.
Granulomatosis with polyangiitis/Wegener's granulomatosis
This is a vasculitis with granulomas and large areas of geographical necrosis occurring in small-sized and medium-sized veins and arteries. There is no gender predilection, but occurs more often in middle-aged patients, although all ages may be affected. Vessels from anywhere in the body may be affected, but there is a preference for upper respiratory tract, lungs and kidneys; GIT involvement is exceedingly rare.
Microscopic polyangiitis
Initially, this form of vasculitis has been confused with a ‘microscopic’ form of PAN or a hypersensitivity vasculitis. In 1994, the Chapel Hill Consensus Conference recognised MPA as a distinct entity despite its overlap with PAN, GPA, EGPA and cutaneous leucocytoclastic vasculitis/angiitis.6 It is a small-vessel (capillaries/venules/arterioles) vasculitis that affects all ages, ethnicities and both genders with almost equal frequency. Very occasionally, medium-sized vessels can be involved by MPA. The GIT is rarely affected, but if present, abdominal pain is the most common symptom.
Although the cause is unknown, MPA is mainly a p-ANCA vasculitis directed against MPO, while c-ANCA against PR-3 is present in 40% of patients.7
Pathology
The usual picture is of a small-vessel vasculitis (sometimes segmental) with numerous eosinophils, fibrin and leucocytoclasia (figure 2A–D). Occasionally, it can mimic PAN and cause a necrotising arteritis of medium-sized vessels. There are few or no immune complexes in the vasculitis vessels.

(A–D) Microscopic polyangiitis of small mesoappendiceal arterioles, featuring eosinophils and leucocytoclasia.
The differential diagnosis (see table 2) includes immune complex-mediated small-vessel vasculitis (Henoch-Schonlein purpura or cryoglobulinaemic vasculitis). The paucity of immune complexes in MPA separates it from the aforementioned vasculitides. MPA is distinguished from GPA and EGPA by the absence of granulomas, peripheral blood eosinophilia and asthma.
Clinical and immunological distinguishing features of the major vasculitides
Immune complex vasculitis
Henoch-Schonlein purpura/IgA vasculitis
The pathogenesis of IgA vasculitis (IgAV) has been linked to immunisation, food allergies, infections (group A––haemolytic Streptococcus and several other infectious agents) and drugs (several, including quinidine, ranitidine, clarithromycin). It has been postulated that patients who develop IgAV have abnormally glycosylated IgA1.1 IgA production recruits inflammatory mediators with activation of complement cascade. The disease may be systemic or limited to the kidney. Typically, it is a self-limiting disease that affects children more than adults, occurring after an upper respiratory tract infection. The usual manifestations occur in the skin (purpura), joints (arthritis), kidney and GIT. The GIT may be the first organ affected in about 14% of cases.8 GIT symptoms are protean and occur in 50%–85% of patients with IgAV. These include diarrhoea, vomiting, diffuse colicky pain, obstruction, perforation, distention, intussusception, haematemesis, melaena, haematochezia and intussusception.3
Pathology
IgAV can affect any part of the GIT, but the second part of the duodenum is preferentially affected. IgAV causes a leucocytoclastic vasculitis of small-sized and medium-sized capillaries, veins and arteries, with the deposition of IgA and C3 in the vessel walls. The differential diagnosis includes the major vasculitides as outlined in table 2.
Cryoglobulinaemic vasculitis
This is a small-vessel vasculitis involving capillaries, venules and arterioles associated with serum cryoglobulins. Ninety per cent of cases are associated with hepatitis C, in which the vasculitis is best called hepatitis C-associated cryoglobulinaemic vasculitis. When the aetiology is not known, then the cryoglobulinaemic vasculitis is regarded as ‘idiopathic’ or ‘essential’.1
Three types of cryoglobulins recognised are as follows:
Type 1: monoclonal, not associated with Rh factor, associated with lymphoma, Waldenstrom's and myeloma
Type 2: monoclonal with Rh factor, associated with lymphoproliferative diseases, rheumatic disease and chronic infections;
Type 3: polyclonal with Rh factor, associated with rheumatic disease and chronic infections
Types 2 and 3 are most frequently accompanied by a vasculitis.
Systemic lupus erythematosus
Pathogenesis: defective suppressor T cells/activate B cells deposition of immune complexes in capillaries. GI symptoms include peritonitis, pancreatitis and a GI vasculitis resulting usually in abdominal pain.
Pathology
Small-vessel and medium-vessel arteritis/venulitis with fibrinoid necrosis in severe cases, acute and chronic inflammation with immune complex and C3 deposition in the media and adventitia layers of the vessel wall.
Mixed connective tissue disease
This shares overlapping features with SLE, scleroderma and polymyositis. These patients have high titres of anti-U1 ribonucleoprotein antibodies, which are thought to be pathogenic. GI symptoms include dysphagia, heartburn, perforation and malabsorption.
Drug-induced vasculitis
Drug-induced vasculitis may be a complication of therapy as a result of prior use of certain medications. It can occur as a systemic syndrome in a minority of patients treated with a drug over a long period of time or more frequently as a cutaneous vasculitis, usually 7–21 days after the drug was first administered.9 The GIT is usually involved as part of systemic involvement rather than in isolation and is also very rarely the initial organ of presentation. Early termination of the offending drug usually results in a complete recovery, as opposed to a more advanced state of disease with a longer drug exposure which may require immunosuppressive therapy.
Pathogenesis
The pathogenesis of drug-induced vasculitis is unclear, but is likely to be multifactorial. It presents with the same clinical picture as patients with an autoimmune profile, suggesting a common mechanism. Drugs contain low molecular weight substances, require the formation of a complex to stimulate antibody formation, and thus elicit an immune response.
Activated neutrophils in the presence of hydrogen peroxidase release MPO from their granules, which convert drugs into cytotoxic products; drugs and their metabolites are immunogenic for T cells, which in turn activat B cells to produce ANCA.
p-ANCA directed to MPO play a key role in the pathogenesis of drug-induced vasculitis, although other factors may be involved such as neutrophil cytoplasm granules, cathepsin G and lactoferrin. Eighty to ninety per cent of cases are positive for p-ANCA, and almost all the patients with drug-induced vasculitis have antibodies to MPO rather than PR3. As for the ANCA-negative drug-associated vasculitis, drugs and their metabolites may accumulate within neutrophils, bind to MPO modifying its configuration, causing an autoimmune response to other autoantigens rendering neutrophil proteins (including elastase, lactoferrin and nuclear antigens) immunogenic.
The presence of ANCA with specificity to more than one lysosomal antigen combined with the presence of antibodies to histones and β-2 glycoprotein I is characteristic for drug-induced vasculitis. There is evidence that these antibodies are pathogenic in small-vessel vasculitis. The presence of multiantigenicity may help to distinguish drug-induced vasculitis from primary ANCA-associated vasculitis.
Genetic factors may also predispose to develop drug-induced vasculitis.
Clinical manifestations
The clinical manifestations of drug-induced vasculitis are non-specific and similar to a primary vasculitis, including less specific syndromes restricted to single organ or tissue involvement. It is hypothesised that more specific organ involvement is subsequent to prolonged drug exposure. Kidney, skin and lung are the specific targeted organs most frequently involved.
Drug-induced vasculitis tends to have a less dramatic clinical evolution compared with idiopathic vasculitis.
Pathology
Drug-induced vasculitis is typically a small-vessel vasculitis with leucocytoclasia and fibrinoid necrosis.
The diagnosis of drug-induced vasculitis is based on the temporal relationship between clinically evident vasculitis and drug administration, with exclusion of other conditions that mimic vasculitis or other types of vasculitis.
A tissue biopsy is usually required to provide a definitive diagnosis of vasculitis and to rule out other diseases/vasculitides. Biopsies are usually taken from skin lesions, kidneys and lungs.
Single-organ vasculitis
This is an ‘isolated PAN-like’ vasculitis in arteries or veins of any size in a single organ with no evidence of systemic vasculitis.1 The vasculitis may be unifocal or multifocal within an organ.
It may represent a forme fruste or limited expression of a systemic vasculitic process. Should the vasculitis declare itself as a systemic disease, then it should be accorded the appropriate nomenclature such as PAN. Clinical, serological and pathological correlation is required to confirm a diagnosis of systemic vasculitis with limited expression in a single organ. Should there be no evidence clinically and/or serologically, then the vasculitis is a solitary organ vasculitis (SOV).
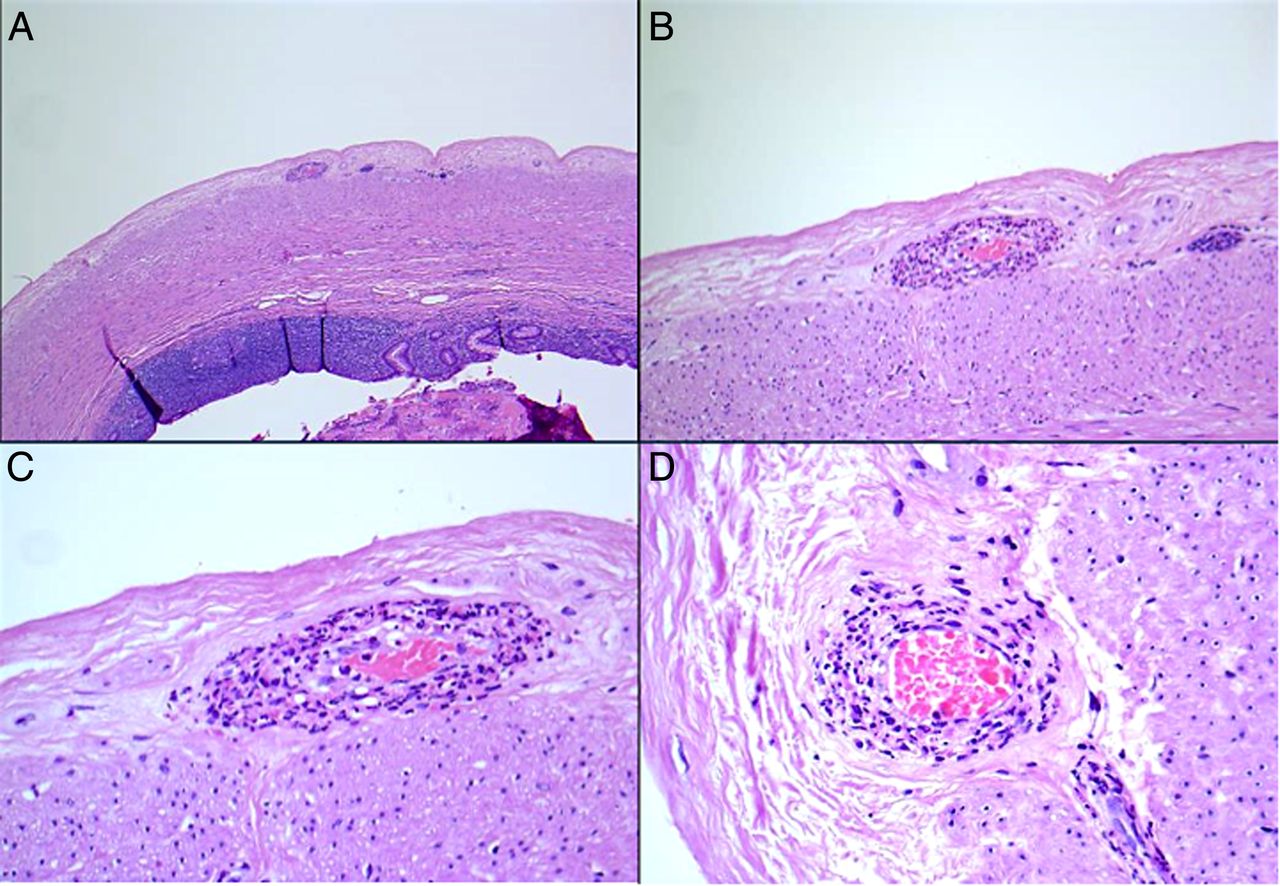
SOV can occur anywhere in the body, and within the GIT, SOV has been reported to occur in several locations within the abdominal cavity, including the oesophagus, stomach, omentum, small and large intestines, appendix, pancreas and gall bladder (the GI site is most frequently the site of SOV).10 The vasculitis can cause GI symptoms such as pain, haemorrhage, infarction, or the SOV may be purely incidental, asymptomatic and occult with the patient presenting with symptoms unrelated to the vasculitis such as tubular adenomas (figure 3A, B), cholelithiasis (figure 4A–D) or even tumours.

(A and B) A tubular adenoma with a solitary organ vasculitis or polyarteritis nodosa-like vasculitis within the lamina propria. Note the fibrinoid necrosis in a small vessel. No accompanying systemic vasculitis was present at the time of polypectomy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
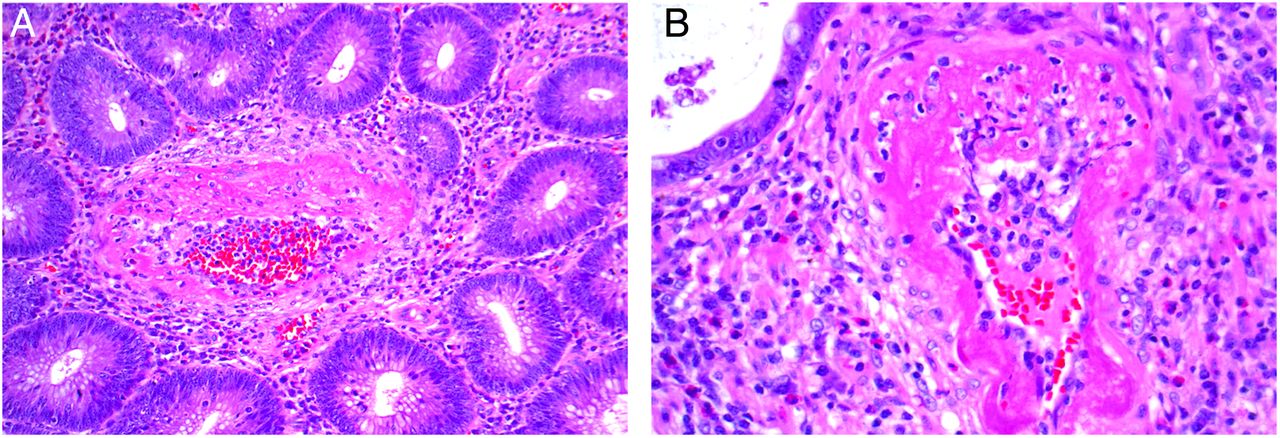
(A–D) Another example of an incidental solitary organ vasculitis involving the wall of the gall bladder that was removed because of gallstone cholecystitis. The patient did not have evidence of systemic vasculitis.
Take home messages
Systemic vasculitides can manifest initially in the gastrointestinal tract.
Look for evidence of vasculitis in specimens (especially in the omentum and deeper layers of the bowel wall) submitted with a history of haemorrhagic infarction or ischaemia.
Multiple sections should be examined if there is a clinical suspicion of systemic vasculitis.
Incidental or solitary organ vasculitis in the absence of systemic vasculitis or symptoms related to vasculitis can be encountered coexisting with GI pathology.
Footnotes
Handling editor Cheok Soon Lee
Contributors RC was responsible for the planning, conduct and writing of the work described in the article. SS contributed to the writing of sections of the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.