Article Text

Abstract
Aims Gene dosage can have a major impact on cell biology, although, hitherto, it has been difficult to study using in vitro models. We sought to refine and accelerate the development of ‘gene dosage’ models through using CRISPR/Cas9 (a gene editing technology) for sequential knockout of gene alleles.
Methods Our method involved (1) using Cas9 nuclease mRNA rather than expression plasmids, (2) using a fluorescently labelled FAM-6 tracr complexed with guide RNA and (3) using high-resolution melting (HRM) analysis to screen for mutations. HCT116 cells, wild-type for TP53, were transfected with different molarities of FAM-6 tracr-labelled and guide RNA targeting different exons of TP53 and selected by fluorescence-activated cell sorting. Single-cell colonies were then isolated, expanded and tested for mutation in the targeted region by PCR/HRM.
Results Out of 32 clones tested, 12 have shown aberrant melting by HRM, giving a targeting efficiency of 37.5%. One clone was sequenced and a heterozygous mutation found — in this case comprising a single base deletion in exon 3. mRNA sequencing confirmed the mutation was expressed, and western blotting for p53 showed the presence of both wild-type and truncated protein bands. Changes in expression of MDM-2 isoforms suggested a functional effect of the induced TP53 mutation.
Conclusions We have developed an in vitro model to study TP53 gene dosage effects. The protocol is efficient and applicable to any gene. Importantly, we have used Cas9 mRNA and labelled tracr/guide RNA to isolate likely mutated cells and HRM for rapid mutation detection.
- P53
- Cancer Genetics
- Molecular Pathology
Statistics from Altmetric.com
Introduction
CRISPR (clustered, regularly interspaced, short palindromic repeat) and Cas (CRISPR-associated) proteins are present in bacteria and archea species. In combination, they form a system that recognises and cleaves foreign nucleic acid derived from phages and plasmids. This system functions as an adaptive immune response to prevent an invading virus from inserting its genome into the host bacterial genome.1 In one of the most transformative technical developments in recent years, this system has been adapted to allow editing of mammalian genes.
Although there are now many different strategies,2 3 the most commonly used system for gene editing is the CRISPR/Cas9 system. This depends on the helicase and endonuclease activity of wild-type Cas9 protein to create mutations at specific target sites by cleaving double-stranded DNA. This is followed by one of two forms of repair (1) non-homologous end joining repair — an error-prone process that is characterised by frequent insertion or deletion of nucleotides resulting in frameshift mutations and early stop codons,4 or (2) homology-directed repair, in which an exogenous DNA template is used to induce missense mutations.5
Cas9 can be directed to cleave any gene of interest in eucaryotic cells by introducing a synthetic guide RNA (sgRNA) sequence. The sgRNA has two components, including a CRISPR RNA (crRNA), which contains sequences complementary to (and thereby identifying) the target gene and adjacent short sequences called protospacer adjacent motif.6 The second component is a sequence called transacting CRISPR RNA (tracr-RNA), which is essential for activating the Cas9 protein. By pooling different guide RNA/Cas9 complexes, a multiplex editing of target loci can be achieved.7 The sgRNA and Cas9 can be packaged into a plasmid, and gene editing can begin by transfecting the construct into cells. The successfully transfected cells need to be identified, and this can be done with a fluorescently tagged Cas9 protein. These cells are then grown as single cell colonies and then they need to be tested for the induction of mutation. This is most commonly done using PCR followed by enzymatic cleavage of heteroduplexes, which naturally form when mutant and wild-type gene sequences are coamplified. Once a clone has been identified to contain the required mutation, its cells can then be used to target the other allele.8
One of the disadvantages of CRISPR/Cas9 gene editing is the possibility of inducing additional off-target genetic modifications, that is, mutation of a different gene. This could confound experimental work and would limit use of CRISPR/Cas9 for therapeutic purposes.9 Off-target mutations could be reduced through limiting the time frame for the activity of Cas9 by transfecting Cas9 mRNA (or its ribonucleoprotein) into the target cells instead of Cas9-expressing plasmid DNA. By electroporating Cas9 ribonucleoprotein directly, several groups have been able to efficiently induce RNA-guided engineered nucleases activity,10 11 and Kim and colleagues have suggested that, in principle, Cas9 protein can be replaced with Cas9 mRNA, which is easily transcribed but not integrated into the host genome.12 Cas9 transcript could still be detected 96 hours post-transfection of Cas9-expressing plasmid DNA, whereas it was absent for the same time point in the case of Cas9 mRNA transfections.13 Liang and colleagues have also reported reduced rates of off-target mutations produced by using Cas9 mRNA and ribonucleoprotein versus Cas9-expressing plasmid DNA.14
In this study, we sought to develop a protocol for gene dosage studies. We have employed Cas9 mRNA because (1) it is easier to transfect into the cells than plasmids, (2) it is translated rapidly into Cas9 protein in the cytoplasm (thereby obviating the need for nucleofection) and (3) it is rapidly degraded, thereby reducing the duration of action of Cas9 and reducing the risks of both biallelic mutation and off-target gene editing. Additionally, a FAM-6-labelled tracr was complexed with guide RNA and Cas9 mRNA to allow for flow-assisted cell sorting of successfully transfected cells. Finally, we used high-resolution melting (HRM) to screen for mutations as it is quick and could be used for screening for mutations in both the first round and the second round of targeting. TP53 was chosen as a target gene to test the feasibility of Cas9 mRNA-based CRISPR as it is an important tumour suppressor gene.15
Materials and methods
Cell culture
HCT116, a colorectal cancer cell line previously shown to be wild-type for TP53, 16 was obtained from the American Tissue Culture Collection. It was cultured in high-glucose Dulbecco’s modified eagle medium (DMEM) (Gibco, 41 965–062) supplemented with 10% fetal bovine serum, 2 mM L-glutamine (Sigma Aldrich, St Louis, Missouri, USA) and maintained in a humidified incubator at 37°C in a 5% CO2 atmosphere. Cells were regularly passaged at 70%–80% confluency and experiments performed up to passage number 20.
Transfection
Lipofectamine MessengerMAX (Invitrogen, LMRNA001) was employed as a transfection reagent. During the transfection cells were maintained in Opti-MEM I Reduced-Serum Medium (Gibco, 31985062).
Wild-type Cas9 mRNA, capped and polyA-tailed, at a concentration of 500 ng/µL was purchased from Sigma Aldrich. Guide RNAs including crRNAs designed to target the first three exons of TP53 (figure 1) as well as the FAM6 labelled and unlabelled tracr RNA oligos were kindly supplied by Dr Gurpreet Balrey (Merck, Middlesex, UK).
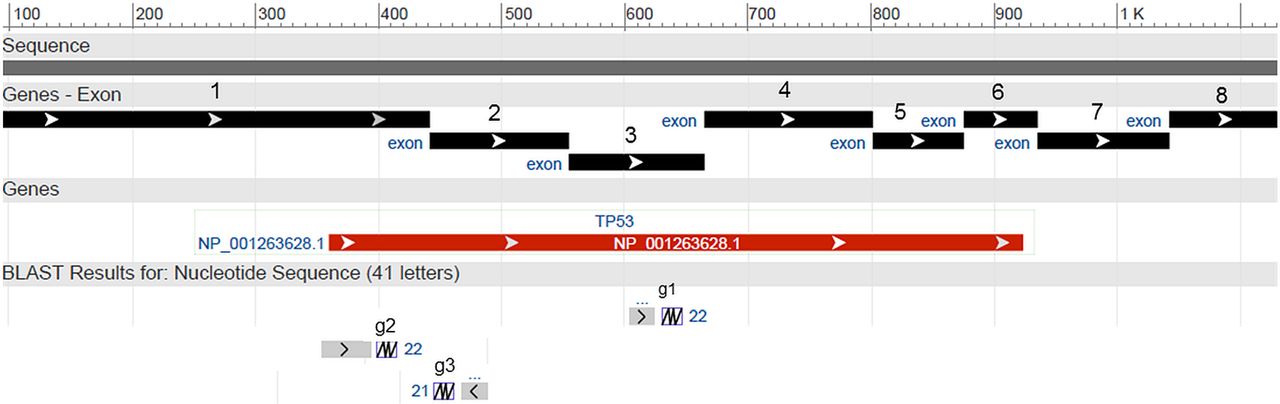
sgRNA (g1, g2, g3) targeting exons 3, 1 and 2, respectively, of TP53 (gi|383209646:5001-24149 Homo sapiens tumour protein p53 (TP53), RefSeqGene (LRG_321) on chromosome 17 genomic coordinates: exon 3: 18311–18329; exon 1: 17440–17458; exon 2: 17589–17608. Guide RNA 3 aligns with the reverse complement sequence of shown exon 2, whereas g1 and g2 align with 5'-3' sense chain. sgRNA, synthetic guide RNA.
Cells were seeded in 24-well plates at a density of 1.2×105 cells/well and left to adhere overnight in a humidified incubator at 37°C, 5% CO2. Transfection was initiated at 70% confluence. The growth medium was changed from DMEM to Opti-MEM Reduced-Serum Medium 1 hour before transfection. Transfection was performed using a mixture of 500 ng per well of Cas9 mRNA, 8, 16 or 24 pmol of three different guide crRNAs (table 1), which were designed to target three different exons of TP53. Equimolar quantities of crRNAs and tracrs (unlabelled tracrs were used as a negative control) were complexed for 5 min in 25 µL of Opti-MEM. Separately, 1.5 µL of Lipofectamine MessengerMAX was complexed with 25 µL Opti-MEM for 5 min. The diluted Cas9 mRNA and sgRNA (constituted by complexed crRNA and tracrRNA) were added to diluted Lipofectamine MessengerMAX, gently mixed and incubated for 10 min at room temperature, while protecting it from light. The mixture was added drop-wise to each well and each experiment was performed in triplicate.
Guide RNA sequences, respective genomic coordinates, target exons referring to transcript variants 5, 6 and 7 of human TP53 and FAM-6-labelled tracr sequence
The cells were incubated in a humidified incubator at 37°C, 5% CO2, and after 6 hours Opti-MEM was replaced with complete DMEM, 10% fetal bovine serum (FBS), 2 mM L-glutamine and incubated for 72 hours.
Flow cytometry and single cell colony formation
At 72 hours post-transfection, cells were dissociated with 0.25% Trypsin-EDTA for 3–5 min at 37°C, resuspended in 10% fetal bovine serum (FBS) DMEM, followed by three washes inphosphate buffered saline (PBS) after after a centrifugation step (1500 rpm, 5 min). Fluorescence-activated cell sorting (FACS) was performed using a Beckman Coulter MoFlo XPD, on the FL1 channel, which was compatible with detection of the FAM-6 fluorophore (excitation at 496 nm, emission 516 nm). Cells in which transfection was performed with sgRNA containing unlabelled tracr were used as a negative control of background fluorescence levels. The pools of cells transfected with 16 pmol sgRNA3, and 24 pmol sgRNA1 and 2, were chosen for sorting, and were resorted to obtain a nearly 100% positive FAM-6 positive cell population. The final sorted cells were first grown to create a stock that was cryopreserved and also to collect conditioned media of each pool of sgRNA sorted cells, resuspended at a density of approximately 10 cells/mL and 100 μL was pipetted into each well of a 96-well plate. This equates to seeding 1 cell per well. Two 96-well plates for each sgRNA were seeded with approximately one cell per well to allow the isolation of single cell clones. Each well was supplemented with 100 µL of conditioned media (48 hours) collected from the respective pool of sorted cells to enhance the formation of single colonies. The single colonies that formed in a 96-well plate were then expanded successively into larger capacity plates (24-well plates, 6-well plates) and ultimately T25 flasks. At this point, each of the expanded clones was split for DNA extraction and cryopreservation in a solution of 10% dymethyl sulfoxide (DMSO), 90% fetal bovine serum.
PCR for HRM analysis of single cell clones
Genomic DNA was extracted from cell pellets of each clone using GenElute Mammalian Genomic DNA Miniprep Kit (Sigma Aldrich, G1N70) following the manufacturer’s recommendations. The eluted DNA was quantified using NanoDrop 2000 (ThermoFisher Scientific) and diluted to 20 ng/µL.
By using primers flanking the regions targeted by the three sgRNAs (figure 1), PCR was performed in triplicates for subsequent HRM analysis. Briefly, 40 ng of genomic DNA template from each clone was added to a PCR mix containing 7.5 µL of 2× PCR master mix hotshot diamond (Clent Life Science, HS002), 1 µL Eva Green dye and 250 nM primer. The PCR reaction was performed in a final volume of 15 µL on a PeqLab primus thermocycler set at 95°C for 5 min, followed by 40 cycles of 95°C for 20 s, 60.5° for 20 s and 72°C for 20 s, followed by an extension step at 72°C for 5 min, 95°C for 2 min for heteroduplex formation and holding at 4°C. The primer sequences used for the PCR are shown in table 2, and their respective positions are illustrated in figure 2.
Map of TP53 intronic and exonic primers used for detection of mutations by high-resolution melting and sequencing.
Primer sequences used for screening and sequencing
HRM was used to screen for mutations through detecting small mutation derived-shifts in the melting temperature of PCR products. HRM has been described as exceptionally sensitive when there are heterozygous mutations as these result in the formation of heteroduplexes, which are very unstable.17 18 PCR products were melted in triplicates by first transferring 10 µL to a 96-well black hardshell plate (Bio-Rad Laboratories, HSP9661) and overlaying with 20 µL mineral oil (Sigma Aldrich, M5904). After a short centrifugation for 5 min at 2500 rpm, the melting was performed on a lightscanner (Idaho Technologies, BioFire Defense) preheated to 62°C. The range of melting temperatures varied with product sizes and sequences analysed, but was set to detect melting peaks from 95°C to 70°C. Exposure was set to ‘Auto’ and data were captured at a ramp rate of 0.1°C/s. The acquired melting data were analysed with the LightScanner Call-IT software V.2.0.0.1.331. Results were viewed as ‘Shifted melt curves’ and ‘Difference curves’ outputs. The criteria for selecting mutant clones were the quality of template DNA, the reproducibility of the aberrant melting patterns on separate PCR runs and the consistency among the three replicates.
RNA extraction, cDNA synthesis and RT-PCR
RNA was extracted from mutant clones and wild-type HCT116 cell line using the total RNA kit (Sigma Aldrich). DNAse I digestion was performed to eliminate contaminant genomic DNA. cDNA synthesis was carried out by using M-MLV transcriptase (Promega, M170A) and 2 µg of RNA was combined with 0.5 µg of hexamer primers (ThermoFisher Scientific, SO142) and up to 12 μL nuclease free water. The mixture was heated to 70°C for 5 min, immediately cooled to 4°C and then the following were added: 5 µL of 5xM-MLV buffer, 1.75 µL of 10 mM dNTPs (ThermoFisher Scientific, R0191), 25 units of RNAse inhibitor (Promega, N2111) and 200 units of M-MLV reverse transcriptase and nuclease free water to make a final volume of 25 µL. The mix was then incubated in a thermocycler for 60 min at 37°C, after which the cDNA was stored at −20°C.
RT-PCR was performed with 1 µL of cDNA template, 1 unit of AmpliTaq 360 DNA polymerase (Applied Biosystems, 4398818), 1× AmpliTaq buffer, 400 nM of primers (E1F/E4R and E3F/E3R), 200 µM of dNTPs and 1.75 mM of MgCl2. To prevent any secondary structure on the template that could have been caused by heteroduplex formation with the induced mutation, betaine (Sigma Aldrich, B0300-1VL) at a final concentration of 1.3M and 1.3% DMSOPCR grade (Sigma Aldrich, D9170-1VL) were used in the RT-PCR. The cycling parameters were set at 94°C for 5 min, followed by 10 cycles of touchdown annealing temperature, decreasing 1°C/cycle (94°C, 15 s; 65°C–55°C, 30 s; 72°C, 40 s), 30 cycles of denaturation at 94°C for 15 s, annealing at 60°C for 30 s and extension at 70°C for 40 s and a final extension at 72°C for 5 min with storage at 4°C.
Cloning of PCR products
In order to get the highest quality sequencing data, PCR products containing amplified cDNA from exons 1 to 4 was cloned into a pCR2.1-TOPO vector after purification using GenElute PCR Clean-Up Kit (Sigma Aldrich, NA1020-1KT) according to the manufacturer’s instructions.
For the ligation reaction, 46 ng of purified PCR product was added to 10 ng pCR2.1-TOPO vector (Invitrogen, K450001) and 1 µL salt solution (1.2 M NaCl, 0.06 M MgCl2) and incubated for 20 min at room temperature. Next, 2 µL of the ligation mix was added to a 50 µL phial of One Shot Chemically Competent E. coli (TOP10, Invitrogen, K450001), gently mixed, incubated on ice for 30 min and then heat shocked for 30 s at 42°C, then quickly returned to ice for 5 min. Next, SOC medium (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20 mM glucose) was added and the mixture incubated for 1 hour at 37°C, shaking at 230 rpm before spreading onto prewarmed selective plates of Luria Broth (LB) agar containing 100 µg/mL of ampicillin (Invivogen, fas-am-s), coated with 40 µL of 40 mg/mL X-gal in dimethylformamide. After overnight incubation at 37°C, 12 white colonies were picked, inoculated in 5 mL of LB containing 100 µg/mL ampicillin (Invivogen, fas-am-b), incubated for 16 hours at 37°C, and then used for plasmid DNA extraction following the instructions on the GenElute Plasmid Miniprep Kit (Sigma Aldrich, PLN350).
Sequencing and analysis
The purified PCR products were diluted to 10 ng/µL and cloned plasmids were diluted 100 ng/µL in nuclease free water. Sanger sequencing was performed at the DeepSeq facilities in the School of Life Sciences at the University of Nottingham using dye terminator chemistry (BigDye V.3.1) on the 3130xl ABI PRISM Genetic Analyzer. The obtained sequences were analysed with BioEdit software V.7.2.5 and aligned to human genomic or transcript TP53 sequences using Mega software V.7.0.1.8. Translation of gene sequences of wild-type TP53 isoform A (NCBI RefSeq: NG_017013.2, Homo sapiens tumour protein p53 (TP53), RefSeqGene (LRG_321) on chromosome 17) and mutant clone G11A8 with a point deletion was performed using the online translation tool ExPASy (http://web.expasy.org/translate/), and the respective protein alignments were performed on Mega software V.7.0.1.8.
Western blot
Wild-type and clone G11A8 HCT116 cells seeded at a density of 106 cells/well were incubated overnight and then lysed using ice-cold radioimmunoprecipitation assay buffer (ThermoFisher Scientific, 10017003), incubated for 10 min on ice with 1× Halt protease and phosphatase inhibitor (ThermoFisher Scientific, 78440) and then centrifuged for 30 min at 16 000× g and 4°C. The supernatant was collected and quantified by BCA assay (ThermoFisher Scientific, 23225). Thirty micrograms of protein were mixed with 4× lithium dodecyl sulfate sample buffer (Invitrogen, 11549166), loaded into a 4%–12% Bis-Tris NuPAGE Gel (Invitrogen, NP0321BOX) and run at constant voltage of 150 V for 90 min. Protein was transferred onto a nitrocellulose membrane using a semidry transfer system (Bio-Rad Laboratories, Turbo-blot), which was incubated in blocking buffer (5% skimmed milk in tris buffer saline (TBST; pH 7.6, 0.1% Tween 20)) for 1 hour and then hybridised overnight at 4°C with mouse monoclonal anti-human TP53 antibody at 1:200 dilution (DAKO, clone do-7, M700101-2) and mouse double minute 2 homolog (MDM2) at 1:500 dilution (Abcam, ab3110), diluted in blocking buffer. Mouse anti-human beta-actin (1:2000 dilution in blocking buffer, Sigma Aldrich, A5441) was used as a loading control and the membrane was incubated for 2 hours at room temperature. After 3× washes of 5 min each in TBST, the membranes were incubated for 1 hour at room temperature with secondary horseradish peroxidase -conjugated rabbit anti-mouse IgG (Sigma Aldrich, A6154), diluted 1:1000. After another 3×5 min washes with TBST and incubation with ECL prime (GE Healthcare, RPN2236) for 5 min, the membranes were scanned with a digital scanner (LI-COR, C-digit) to detect chemiluminescence.
Results
Identification of targeted cell pools
By using flow cytometry to detect FAM-6-labelled tracr transfected in the cells, we were able to detect and identify the molarities showing best FAM-6 labelling in the population of cells successfully transfected with sgRNA containing FAM6-labelled tracr and Cas9 mRNA (figure 3). The labelled population ranged from 1.67% to 4.55% in the pooled triplicates containing over 3×106 cells. The sorted cells were enriched in a second sorting, increasing FAM-6 positivity purity from the initial 50%–80% obtained in the first sorting (figure 4).

Detection of HCT116 cells transfected with a mix of 16 pmol (top panel) or 24 pmol (lower panel) of FAM-6- labelled sgRNA and 500 ng Ccas9 mRNA. First plot on the left shows the forward scatter (FSC) and side scatter (SSC). Second plot shows fluorescence (FL-1 channel) in cells transfected with unlabelled sgRNA (negativive control). Remaining plots show labelling percentage in cells transfected with sgRNA 1, 2 and 3 complexed with FAM-6- labelled tracr. Transfected cells marked with an asterisk were selected for sorting of the complete cell pool. sgRNA, synthetic guide RNA.

Enrichment of transfected cells. The purity of the labelled population was increased in a second sorting where 51.14%–-83.08% of cells from the first sorting round where effectively selected to continue expansion and isolation of single cell colonies. FACS, fluorescence-activated cell sorting.
Isolation of single cell clones
In total, 32 colonies were selected for further expansion, genomic DNA extraction and cryopreservation. The three pools of selected sgRNA1, sgRNA2 and sgRNA3 initially targeted cells were also cryopreserved to allow a repeated selection of single cell clones in case a mutant clone could not be detected.
Detection of mutant clones by HRM
HRM was used to screen for the presence of mutant clones isolated from the pools of sgRNA 1, 2 and 3 targeted cells. The use of normal template genomic DNA from untargeted HCT116 in the PCR reactions served as a baseline control to which shifts in melting temperatures could be compared. Of the 32 clones that were tested, 12 (37.5%) showed aberrant melting (figure 5).

HRM- based screening for TP53 mutation. Data from cClone G11A8 (red trace) and the wild-t type containing parent HCT116 (grey). Normalised and temperature- shifted melting curves are shown in the upper panel and demonstrate a left shift in G11A8 compared withto parental cell line indicating the presence of a mutation. This is confirmed in the difference plot shown in the lower panel. FACS, fluorescence-activated cell sorting; HRM, high-resolution melting.
Confirmation of indel mutation and its expression by sequencing
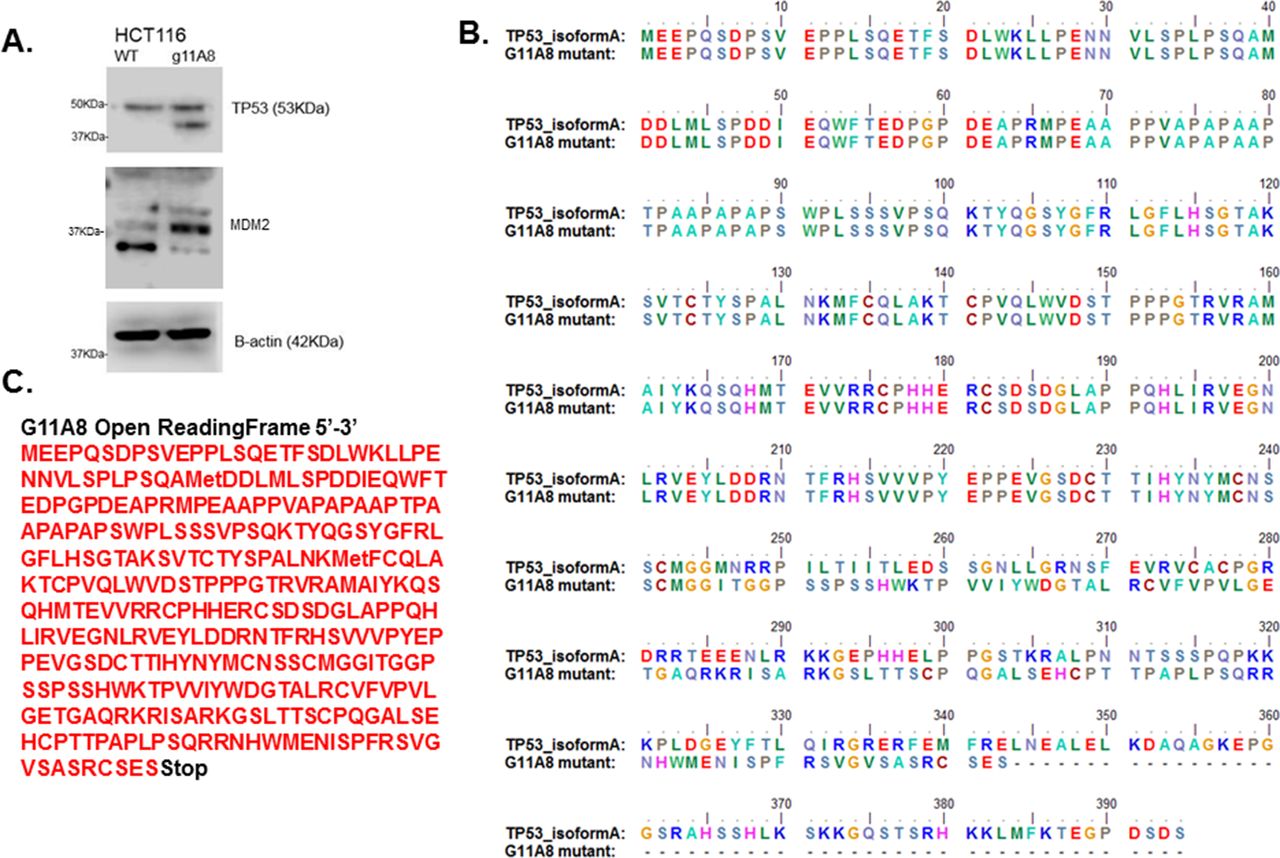
HRM is a robust method for mutation detection and one clone, G11A8, was selected for further expansion and testing. In order to confirm that mutation, mRNA was extracted, reverse-transcribed and the coding sequence spanning exons 1–4 of TP53 was cloned and sequenced. Analysis of the sequencing data showed that the selected clone contained both wild-type and mutant sequence, thereby confirming the heterozygous nature of the mutation (figure 6). The mutation consisted of a single G deletion within the region complementary to the targeting sequence of sgRNA1 — CTGCATGGGCGGCATGAAC (deleted base underlined). This corresponds to the mutation c735delG in the TP53 coding sequence. Usually frameshift mutations (insertions or deletions that are not a multiple of three) result in a truncated protein. The new sequence created as a consequence of the c735delG mutation, when translated, is predicted to produce a truncated protein of 343 amino acids with a molecular weight of 41 kDa (figure 7C).

Direct sequencing of TP53 eExon 3 mRNA. In order to confirm expression of the mutation, mRNA encoding exons 1 – 4 were cloned and sequenced. Some clones contained wild- type sequence (top electropherogram), while whilstother clones contained mutant sequence comprising a single base deletion (bottom electropherogram). The arrow marks the deleted base. FACS, fluorescence-activated cell sorting.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A). Presence of a truncated TP53 protein form and different expression of MDM2 isoforms in the mutant TP53 cell line. (B). Alignment of wild-type TP53 isoform A and G11A8 mutant translated protein sequences. (C). Open-reading frame of mutant G11A8.
Confirmation of truncated TP53 protein and its functional effect
Having confirmed that the mutation was present at the mRNA level, we sought to confirm that the mutation led to a truncated protein form. A western blot with protein lysates from both the parental cell line HCT116 (wild-type for TP53) and mutated clone G11A8 was performed. The parental cell line contained a single band of correct size (approximately 53 kDa). The mutated clone showed both the wild-type band and an extra band, corresponding to the predicted truncated TP53 protein of approximately 41 kDa (figure 7A). To confirm that there was a functional consequence of the induced mutation, we performed a western blot for MDM2. This protein has a complex relationship with p53 as it is both a target of p53 and an inhibitor of p53. Comparison of the parental HCT116 with the mutated clone G11A8 confirmed that there was indeed a functional effect with a change in the pattern of MDM2 splice variants in the mutated clone (figure 7). When aligning the translated wild-type coding sequence of human TP53 isoform A ((NG_017013.2:15 957–16030, NG_017013.2:16 148–16169, NG_017013.2:16 279–16557, NG_017013.2:17 315–17498, NG_017013.2:17 580–17692, NG_017013.2:18 261–18370, NG_017013.2:18 714–18850, NG_017013.2:18 943–19016, NG_017013.2:21 836–21942, NG_017013.2:22 861–22942) Homo sapiens tumour protein p53 (TP53), RefSeqGene (LRG_321) on chromosome 17) with the respective G11A8 presenting point deletion on the gRNA1 target site (CTGCATGGGCGGCAT[G]AAC), a significant change in the protein can be observed from the point mutation occurring. When a guanine (G) is deleted in the ATG codon, which would translate into methionine in the wild-type protein, a frameshift occurs to synthesise isoleucine (a.a. 246) (figure 7B) and missense amino acids follow, until a stop codon is produced.
Discussion
In cancer biology, changes in gene dosage may have a significant effect on cell biology. Oncogenes such as β-catenin depend on ‘just right’ signalling for maximum activity19; tumour suppressors, such as TP53, would be expected to confer some selective advantage following somatic mutation (prior to loss of heterozygosity) and some tumour suppressors, such as PARK2, appear to mediate effect through haploinsufficiency.20 We have sought to develop an efficient protocol to establish models for gene dosage studies by taking advantage of recent technical developments in gene editing and combining these with FACS and HRM to enrich for the target population and screen for mutations. Using this methodology, we have induced a heterozygous truncating mutation in TP53 and shown that it causes a change in the pattern of MDM2 expression.
The first step in our method involved introduction of the sgRNA and the Cas9 mRNA into the cells through lipid-based transfection. This would result in a pulse of gene editing activity (lasting until the mRNA was degraded) rather than the constant editing activity, which may be expected if inducing CRISPR/Cas9 machinery from a DNA plasmid expression vector. Therefore, the main advantage of this methodology is a reduction of off-targets gene editing as the translated Cas9 protein has a short half-life and reduced editing time.14
After single cell cloning, the next step was to screen the clones for the presence of somatic TP53 mutation. Rather than performing genomic cleavage detection (GCD) on targeted cells, we used HRM with stringent criteria to identify the presence of single mutant clones in our methodology. HRM has a number of advantages over GCD. HRM is a cheap, simple and very sensitive method for mutation screening, performed in a closed tube, immediately after PCR.21 In contrast, GCD requires PCR products to undergo an enzymatic digestion followed by resolution on an agarose gel in order to identify cleaved PCR products. Another advantage of HRM is that each heteroduplex has a unique melting pattern, and thus if there is a second round of CRISPR/Cas9 editing to produce a homozygous mutant, HRM can be used to distinguish colonies with double mutations from single mutations. In contrast, GCD could not do this since the mismatches leading to heteroduplex formation will be approximately in the same position whether single or double mutations, and this will lead to cleavage fragments of the same size.
Aberrant melting patterns were observed for 37.5% of the clones. Depending on the cell line and transfection method, this is commensurate with the range of efficiencies found by Liang et al targeting HPRT using the Cas9 mRNA format in several established and primary human cell lines (0%–70%), even though efficiency of gene editing in HCT116 cell line was not tested.14 However, in the study by Liang et al, the induced mutations were not tested in individual single cell colonies but were inferred from PCR product cleaved fragment density from PCR performed on pooled colonies; therefore, our results are not in comparable terms.
In summary, we have combined several methods to develop a CRISPR/Cas9-based system for creating models for studying gene dosage effects. The methodology was used to create a heterozygous truncating mutation of TP53, which resulted in changes in MDM2 expression. Our protocol is rapid; it will probably reduce off-target effects, and FACS enrichment allows isolation of a single cell clone with a high probability of mutation and HRM allows mutation detection in both the heterozygous and the homozygous state.
Take home messages
A gene dosage model was developed by applying a variant of the CRISPR/Cas9 method involving the use of Cas9 mRNA and FAM6- labelled sgRNA.
High-resolution melting (HRM) was applied to screen for mutant clones instead of enzymatic genomic cCleavage dDetection.
A TP53 heterozygous truncating mutation was created, resulting in changes in MDM2 expression.
Acknowledgments
The authors would like to thank the technical assistance of Dr Matthew Carlile of the University of Nottingham DNA Sequencing Laboratory, School of Life Sciences, and Nicola Croxall at the Flow Cytometry Facility, School of Medicine, Queen’s Medical Centre, Nottingham, as well as the support of Dr Gurpreet Balrey (Merk, UK).
References
Footnotes
Handling editor Runjan Chetty.
Contributors TPR performed the experimental work, interpreted data and drafted the manuscript. HOE contributed to data analysis and revisions of the manuscript. MI contributed to the experimental design and critically revised the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.