Article Text

Abstract
Fibroadenomas of the breast are benign fibroepithelial tumours most frequently encountered in women of reproductive age, although they may be diagnosed at any age. The fibroadenoma comprises a proliferation of both stromal and epithelial components. The mechanisms underlying fibroadenoma pathogenesis remain incompletely understood. In the clinical setting, distinguishing cellular fibroadenomas from benign phyllodes tumours is a common diagnostic challenge due to subjective histopathological criteria and interobserver differences. Recent sequencing studies have demonstrated the presence of highly recurrent mutations in fibroadenomas, and also delineated the genomic landscapes of fibroadenomas and the closely related phyllodes tumours, revealing differences at the gene level, which may be of potential adjunctive diagnostic use. The present article provides an overview of key studies uncovering genetic and genomic abnormalities in fibroadenomas, from initial karyotype reports revealing myriad cytogenetic aberrations to next-generation sequencing-based approaches that led to the discovery of highly recurrent MED12 mutations. A thorough understanding of these abnormalities is important to further elucidate the mechanisms by which fibroadenomas arise and to refine diagnostic assessment of this very common tumour.
- cancer
- genetics
- breast pathology
- molecular pathology
Statistics from Altmetric.com
Introduction
Breast fibroadenomas (FAs) are common, benign fibroepithelial lesions composed of stromal and epithelial components, with intralobular stroma enclosing glandular spaces lined by luminal epithelium and myoepithelium. This tumour is most frequently encountered in adolescent girls and young women within the first three decades of life, although it may also be diagnosed at any age.1–3 FAs have been clinically observed to be hormone dependent—stimulated by oestrogen, progesterone and lactation during pregnancy, as well as shrinking during menopause.4–6 Typically, they present as slow-growing, firm and solitary nodules, although synchronous and metachronous multifocal lesions have also been described in the literature.7 While FAs are benign neoplasms, one study suggested that they are an independent risk factor for breast cancer,8 while another report provided evidence that they are associated with a twofold increase in relative risk of developing invasive breast carcinoma after 20 years.9 Although the genetic risk factors for FAs are currently unknown, patients with Carney complex may develop bilateral or multiple myxoid FAs, and bilateral and/or multiple FAs have been significantly associated with family history.10 11 In renal transplant patients, the use of cyclosporin has been linked to the development of bilateral and/or multiple FAs.12–14 In this review, we appraise the literature on the genetics and genomics of breast FAs.
Cytogenetic aberrations
In the early 1990s, structural abnormalities including rearrangements of chromosomes 1, 6, 7, 8, 12 and 15 were described in isolated case reports of breast FAs, together with monosomy of chromosomes X, 12 and 21 and trisomy of chromosomes 5, 6, 11, 17 and 20.15–20 Of particular interest was the 12q13–15 region, which was also implicated in several other benign tumours in different anatomical sites such as lipomas, uterine leiomyomas and pleomorphic adenomas.21–23 Non-random involvement of alleles found in chromosomes 1 and 17 were previously reported in breast cancer,24–31 and it was thought that there could be overlapping mutations driving the development of FAs as well.32 However, no consensus could be achieved regarding specific mutations that result in the development of FAs.
This changed in 1995 when Schoenmakers et al identified a candidate gene named HMGI-C within the multiple aberration region on chromosome 12q15 that frequently harbours breakpoints in benign solid tumours, which was mutated in FA together with seven other tumour types.33 In a later clinical case report of an intracanalicular FA showing a clonal chromosomal aberration t(4;12), Staats et al used fluorescence in situ hybridisation (FISH) analysis for the chromosomal region 12q14–15 and narrowed down the breakpoint to a 230 kb fragment that belonged to the HMGI-C gene.34 However, this was only shown to be true in a single case of FA, and thus the involvement of this gene was weakly supported at best. Given the technologies of the 1990s, the approaches available to understand the genetics of FAs were largely limited to karyotype studies, positional cloning and FISH analysis, which could not search the entire genome efficiently to pinpoint specific genetic loci involved in FAs.
Microsatellite instability and loss of heterozygosity
As technological advancements in biological assays emerged towards the later part of the 1990s, Deng et al reported that loss of heterozygosity (LOH) was detected in morphologically normal lobules found beside malignant breast tumours, with the most frequent aberration found in chromosome 3p22–25. Of 10 breast carcinomas with this aberration, 6 cases showed the same LOH within normal lobules, suggesting the presence of a tumour suppressor gene at 3p22–25.35 Yet, it was shown by Lizard-Nacol et al that in FAs, there was an absence of many genetic alterations such as gene amplification commonly found in breast carcinomas.36 Combining this with previous studies that reported associations between FAs and breast cancer risk,37–40 McCulloch et al decided to study the incidence of microsatellite instability (MSI) and LOH in FAs and showed that it was low at 11 different loci: TPOX, D3S1514, D3S1675, FABP, D4S243, D9S254, TH01, D13S289, SCA3, D17S559 and D17S855.41 Additionally, epithelial cells in a single case of FA in a series of benign breast diseases preceding invasive breast cancer were shown to be negative for LOH at chromosome 3p.42 The markers used for this subsequent study included D3S1766, D3S1612, D3S1244, ITIH-1, D3S4102 and LUCA2.1. Later reports, such as by Franco et al, also verified that LOH was indeed a rare feature in FAs,43 as opposed to phyllodes tumours that displayed extensive LOH on genome-wide single-nucleotide polymorphism (SNP) array analysis.44 Although more specific regions of the genome could be assayed to search for abnormalities, MSI and LOH were features hardly found in FAs, and hence studies focusing on them generated little knowledge in terms of the pathological mechanisms that underlie FAs.
TP53 and ras mutations
The unequivocal importance of the tumour suppressor gene TP53 is clearly demonstrated in its abrogation in over half of all human sporadic cancers, in spontaneous tumour development in Trp53-knockout mice as well as in patients with Li-Fraumeni syndrome who inherit a mutant TP53 allele.45 46 Over the course of tumour development, sporadic TP53 mutations may arise, which are quickly followed by LOH, leading to total p53 deficiency. This accelerates both initiation and progression of tumour growth, and tumours displaying a lack of p53 are often characterised by more aggressive characteristics such as local invasiveness and metastatic ability.47–50
TP53 was first examined in FAs over two decades ago by Koutselini et al who studied expression of this gene in cytological specimens from both benign and malignant breast tumours using a monoclonal antibody (mAb) that detected p53. In 38 out of 39 cases (97.4%), benign epithelial breast cells obtained from FAs, fibrocystic change as well as smears from nipple discharge stained negative for p53.51 This was contradictory to a study 4 years later, which showed that one in eight FAs exhibited a non-silent TP53 mutation.52 This was then opposed by Franco et al who demonstrated that there was an absence of TP53 mutations in FAs from women who remained unaffected by breast cancer over an average follow-up period of 10 years.43 Franco et al went on to demonstrate that TP53 gene mutations were absent in FAs, regardless of whether or not they were diagnosed in women affected by breast carcinoma.53 Lastly, the findings by Schneider et al showed that p53 mRNA and protein levels in FAs were higher compared with adjacent normal breast tissue in women of reproductive age, providing further conflicting evidence about p53 in this disease.54 Taken together, it can be concluded that while the role of p53 has been thoroughly established in human cancers, its implication in the pathogenesis and development of FAs was unproven.
Mutations in the ras oncogenes H-ras, K-ras and N-ras have long been known to contribute towards uncontrolled cell proliferation ultimately leading to tumour development.55 As small GTPases, ras proteins alternate between two states: the inactive guanosine diphosphate-bound and the active guanosine triphosphate (GTP)-bound conformations.56 57 They regulate cell fates through the coupling of receptor activation with downstream effector pathways that elicit cellular responses such as increased proliferation, differentiation and survival.58–60
Only 2 out of 10 (20%) FAs were scored positively when stained using a ras-sensitive mAb, as compared with 19 out of 30 (63.3%) infiltrating ductal carcinomas. Interestingly, these two were multiple FA cases.61 Using a combination of PCR amplification and allele-specific oligonucleotide hybridisation techniques, Kumar et al found that out of seven rat FAs induced by neonatal exposure to nitrosomethylurea, H-ras was mutated in three of them (42.9%), while K-ras was mutated in one case (14.3%).62 Although FAs exhibited ras oncogene activation, it was proven to be within the basal levels of ras activation in normal breast, brain and peripheral nerve tissues.63–65 On a related note, DNA microarray analysis revealed that the Ras homologue enriched in brain (RHEB) protein, also involved in cell growth and differentiation, was expressed at levels more than twofold higher in FAs than in normal tissues. The average RHEB:β-actin ratio of FAs was found to be two times greater than that in infiltrating ductal carcinomas, suggesting RHEB immunohistochemical staining as a possible secondary diagnostic tool to distinguish between the two.66 However, further supporting literature on the role of RHEB in FAs is scant. In summary, the ras oncogenes and related homologues have not been shown to play consequential roles in FAs.
Hormone involvement
Hormones such as oestrin, now more commonly known as oestrogen, were implicated in the development of breast lesions including gynaecomastia and FAs as early as over 80 years ago.67 When nine benign breast conditions including eight fibrocystic lesions and one FA were processed into slices and cultured in medium, the addition of prolactin (PRL) caused increased DNA synthesis as observed by a higher rate of incorporation of radioactive thymidine, elegantly showing PRL as a possible mitogenic stimulant in FAs.68 This stood in contrast to a study by Manni et al using the soft agar clonogenic assay: the only case of FA included in the study did not respond to PRL administration, resulting in no colony formation.69 This was supported by the findings of Kumar et al who observed that 2 cases of FA stained negative for tissue-bound PRL, as opposed to 14 out of 15 cases of apocrine metaplasia of the breast, which exhibited strong PRL staining.70
Compared with normal breast tissue, FAs were also rigorously proven to possess a higher expression level of the prolactin receptor (PRLR) gene by multiple methods including quantitative PCR, immunocytochemistry and immunohistochemistry.71–73 As the role of PRL receptors became gradually established in FAs, it started a hunt for mutations in the gene encoding these transmembrane proteins.
In a study involving 74 patients with multiple fibroadenoma (MFA) and 170 control subjects, Bogorad et al identified four patients harbouring a heterozygous SNP in exon 6 of the PRLR gene, which resulted in a substitution mutation changing isoleucine to leucine at position 146 of the PRLR extracellular domain. This single mutation was enough to confer constitutive activity to the PRLR variant, confirmed by PRL-independent functions of PRLR tyrosine phosphorylation, activation of signal transducer and activator of transcription 5 signalling, transcriptional activity towards a PRL-responsive reporter gene, and cell proliferation and protection from cell death.74 A further study of 95 women with MFA by Courtillot et al from the same research group reported another SNP in exon 5 that encoded the amino acid substitution of isoleucine to valine at position 76 of the PRLR. However, in their study cohort, only 15% of the MFA cases harboured PRLR that showed basal activity in vitro, raising questions about whether PRLR was widely involved in the aetiology of MFAs and whether it is a relevant inhibitory target for these patients.75
The third and definitive study by this group involving another 71 women affected by MFA provided conclusive clinical evidence that gain-of-function PRL receptor variants PRLRI146L and PRLRI76V were indeed not associated with MFA risk. One gripping point raised was that although these variants’ gain-of-function properties were ascertained in reconstituted cell systems, they were not replicable in ex vivo analysis in mammary tissues of mouse or human origin, which suggested that the cellular effect of these variants in vivo was not enough to result in mammary tumour development.76
MED12 mutations
In a landmark study, the use of next-generation sequencing (NGS)-based exome sequencing led to the discovery of highly recurrent Mediator complex subunit 12 (MED12) somatic mutations in FAs of the breast. These mutations were found in 4 out of an initial batch of 8 (50%) FA samples, before being further ascertained in another 54 out of 90 (60%) FAs (figure 1). Out of these 98 FAs sequenced, 41 of them (42%) had point mutations in codon 44 of exon 2 of the MED12 gene.77

Fibroadenoma that on genomic analysis was found to have a single MED12 frameshift deletion. The fibroadenoma shows circumscribed borders and both pericanalicular and intracanalicular growth patterns (A). Higher magnification (B) shows hyalinised stroma around epithelial elements that are stretched into arc-like formations.
The results from this paper were met with multiple studies in quick succession globally, confirming that MED12 mutations are found in up to 65% of FAs, with an overwhelming majority of them being located in codon 44 of exon 2.78–80 Laser microdissection studies have also revealed that the MED12 mutations are limited to the stromal compartment,77 81 with mutation status correlating with stromal but not epithelial protein expression.82 This is supported by the fact that the histological pattern that most frequently harbours MED12 mutations is the intracanalicular FA,79 81 which exhibits stroma growing into and compressing the epithelial compartment, stretching the ducts into elongated, arc-like formations.1 Collectively, this suggests that the initiation of FA development may likely be due to a MED12 mutation event within the stromal compartment, much like the uterine leiomyoma (UL) in which MED12 mutations were first found and have been established as integral for UL tumourigenesis.83 84 In both FAs and ULs, there is a stark absence of recurrent point mutations other than in MED12, which suggests that MED12 mutations might be sufficient for tumourigenesis in both tumour types. However, although there is high mutation frequency and extremely similar mutational patterns in the MED12 gene between FAs and ULs, MED12 mutations were only found in 3 of 41 (7.3%) uterine leiomyosarcomas, a malignant tumour closely related to ULs.85 This contrasts against the persistently higher mutation frequency of MED12 in phyllodes tumours of all grades (although some studies have found a relatively lower frequency of MED12 mutations in malignant phyllodes tumours), the less innocuous fibroepithelial tumour regarded to be closely related to FAs. This suggests that MED12 mutations are key drivers behind the spectrum of breast fibroepithelial tumours from benign FAs to phyllodes tumours.78
Akin to the uterus, the breast is one of the main target sites for oestrogen,86 and given that the Mediator complex has been demonstrated to have cellular interactions with oestrogen receptors α and β,87 MED12 mutations in intralobular stroma may be implicated in aberrant oestrogen signalling that ultimately leads to FA pathogenesis. A recent study by Lozada et al, however, has shown that myxoid FAs lack MED12 mutations, suggesting that this particular variant may arise through mechanisms independent of MED12.88 The interaction of cellular MED12 with Cyclin C has been demonstrated to be dependent on the first 100 amino acids in the primary structure of MED12, including those encoded by the entire exon 2. Somatic mutations in MED12 exon 2 have been shown to adversely affect interactions between MED12 and Cyclin C-CDK8/CDK19, causing loss of Mediator-associated, Cyclin-dependent kinase (CDK) activity, resulting in RAD51B expression and eventual tumourigenesis.89 90 Within the CDK module of Mediator, Cyclin C, CDK8 and MED12 are three subunits that are exclusively recruited in the presence of strong p21 transcriptional upregulation, which occurs downstream of the p53-dependent cell cycle arrest response.91 This further suggests that MED12 exon 2 mutations can be deleterious with regard to p53-dependent cell cycle arrest. Therefore, the tumourigenic impact of somatic MED12 mutations is likely to be multifaceted, involving aberrant oestrogen signalling as well as attenuated cell cycle arrest, and thus warrants further investigations to better understand its full scope.
Other mutations
Previously, somatic missense mutations in RARA were only reported in acute promyelocytic leukaemia that was refractory to treatment, as well as in other solid tumours at low frequencies below 5%.92 Targeted deep sequencing of 21 FAs revealed mutations in the retinoic acid receptor alpha (RARA) gene to be highly clustered within the region that encodes the ligand-binding domain, in 3 (14.3%) of these cases. On top of this, missense mutations were detected in FLNA, PCLO, CHD8 and ROS1, while frameshift mutations were found in MLL2 and PCNXL4.93 Taken together, the identification of recurrent MED12 and RARA mutations within the stromal compartment of FAs points towards clonality. This counters earlier investigations into the clonality of FAs, which used PCR and comparative genomic hybridisation-based approaches to arrive at the conclusion that FAs are polyclonal.94–97 This disparity may be attributed to low stromal cellularity within the FAs that may have masked their true clonality, supported by the fact that FAs with high stromal cellularity or ‘phyllodal features’ were demonstrated to be monoclonal.98 99
Among breast fibroepithelial tumours, mutations in FLNA, SETD2, MLL2, BCOR and MAP3K1 were observed to be more common in phyllodes tumours than FAs. In addition, borderline and malignant phyllodes tumours disclosed mutations in NF1, RB1, PIK3CA, EGFR, TP53 and ERBB4, which were largely absent in FAs and benign phyllodes tumours.93 These genomic differences may be of potential clinical use in distinguishing FAs from phyllodes tumours, an important separation as there are divergent management approaches. It is postulated that similar mutational profiles exist for cellular FAs, which represent a diagnostic difficulty in their distinction from phyllodes tumours, though specific data on cellular FAs remain pending.
Among breast papillary lesions that may occasionally display slight morphological similarity to fibroepithelial tumours, a high prevalence of PIK3CA/AKT pathway mutations has been found.100 There are currently no papers in the literature highlighting frequent mutations of PIK3CA and related genes in FAs. On the other hand, studies describing the presence of MED12 mutations in intraductal papillomas are scant, with one report published this year showing a 0% mutation rate of MED12 in six intraductal papillomas.101 Furthermore, another recent publication demonstrated tubular, lactating and ductal adenomas to lack mutations in exon 2 of the MED12 gene, and that ductal adenomas showed recurrent mutations in GNAS and the PI3K–AKT pathway.102 Ductal adenomas of the breast have been previously postulated to be closely related to or are in fact sclerosed intraductal papillomas.103
Conclusion
Knowledge surrounding FA genetics and genomics has progressed rapidly over the last 20 years—from mostly karyotype studies and FISH analyses at the beginning to the NGS-based approaches today. Table 1 and figure 2 show a summary of the genetic and genomic discoveries in FAs. The next step forward will be to capture their genomic features for correlation with clinicopathological features, which will allow us to evaluate whether genomic data may be harnessed as a secondary adjunct to refine diagnostic assessment of fibroepithelial tumours. In particular, distinguishing cellular FA (figure 3) from benign phyllodes tumour is a common diagnostic challenge, which was brought to greater attention by an interobserver study in which consensus among all pathologists was achieved in only 2 (9.5%) of 21 difficult fibroepithelial lesions. This can be attributed to subjective histopathological criteria such as stromal cellularity, stromal condensation, atypia and well-developed leaf-like growths.99 104 Studies that sought to differentiate FAs from phyllodes tumours using protein immunohistochemistry and other methods have so far not been able to consistently assist in resolving this diagnostic dilemma.105–108 As such, further investigations are warranted to understand whether genomic features can be clinically useful. Finally, a recent study involving massively parallel sequencing analysis and formal clonality analysis of FAs and phyllodes tumours has shown that MED12 mutations are extremely common among them and that a clonal relationship likely exists between FAs and phyllodes tumours that share identical MED12 mutations.109 Therefore, additional studies are also required to shed greater light on the progression of fibroepithelial lesions and whether FAs do constitute the substrate from which phyllodes tumours develop. We are gradually arriving at a more comprehensive understanding of the molecular underpinnings driving the formation of fibroepithelial tumours.

Key genomic and genetic abnormalities reported in breast fibroadenomas. Findings are colour coded according to the type reported: red—cytogenetic aberrations; yellow—microsatellite instability and loss of heterozygosity; blue—TP53 and ras mutations; purple—prolactin receptor mutations; green—MED12 mutations. FA, fibroadenoma; HMGI-C, high mobility group protein; LOH, loss of heterozygosity; mAb, monoclonal antibody; qPCR, quantitative PCR; RHEB, Ras homology enriched in brain; SNP, single-nucleotide polymorphism.
{kind=link}
{kind=link}
{kind=link}
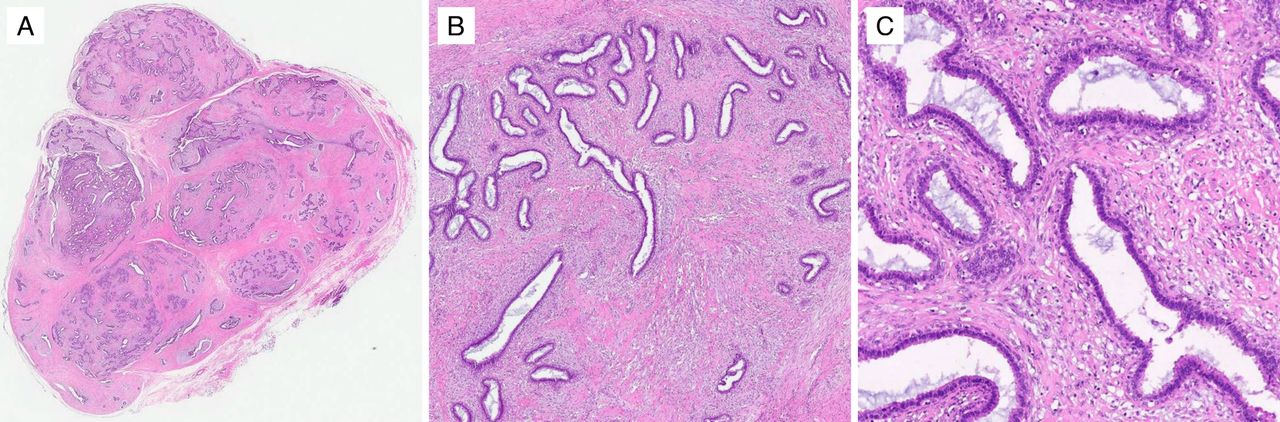
Cellular fibroadenoma at low magnification (A) shows a typical appearance of a proliferation of both epithelial and stromal components. At medium (B) and high (C) magnifications, the increased stromal cellularity is observed. Cellular fibroadenomas may be difficult to distinguish from benign phyllodes tumours when there is a predominant intracanalicular growth pattern.
Key publications on genomic and genetic abnormalities reported in breast fibroadenomas
References
Footnotes
Handling editor Runjan Chetty.
Contributors BNL performed the literature review and wrote the review paper. NDMN took the photographs of the fibroadenoma cases harbouring MED12 mutations. AAT, JYHL, CSL and BTT edited the manuscript. PHT coordinated the above and edited the manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.