
Abstract
Variegate porphyria (VP) is an inherited metabolic disease resulting from the partial deficiency of protoporphyrinogen oxidase, the penultimate enzyme in the heme biosynthetic pathway. We have evaluated the clinical and biochemical outcome of 103 Finnish VP patients diagnosed between 1966 and 2001. Fifty-two per cent of patients had experienced clinical symptoms: 40% had photosensitivity, 27% acute attacks and 14% both manifestations. The proportion of patients with acute attacks has decreased dramatically from 38 to 14% in patients diagnosed before and after 1980, whereas the prevalence of skin symptoms had decreased only subtly from 45 to 34%. We have studied the correlation between PPOX genotype and clinical outcome of 90 patients with the three most common Finnish mutations I12T, R152C and 338G→C. The patients with the I12T mutation experienced no photosensitivity and acute attacks were rare (8%). Therefore, the occurrence of photosensitivity was lower in the I12T group compared to the R152C group (P=0.001), whereas no significant differences between the R152C and 338G→C groups could be observed. Biochemical abnormalities were significantly milder suggesting a milder form of the disease in patients with the I12T mutation. In all VP patients, normal excretion of protoporphyrin in faeces in adulthood predicted freedom from both skin symptoms and acute attacks. The most valuable test predicting an increased risk of symptoms was urinary coproporphyrin, but only a substantially increased excretion exceeding 1000 nmol/day was associated with an increased risk of both skin symptoms and acute attacks. All patients with an excretion of more than 1000 nmol/day experienced either skin symptoms, acute attacks, or both.
Similar content being viewed by others

Introduction
Variegate porphyria (VP (MIM 176200)) is an inherited metabolic disease that results from the partial deficiency of protoporphyrinogen oxidase (PPOX, (E.C.1.3.3.4)), the penultimate enzyme in heme biosynthesis.1 PPOX catalyses the six-electron oxidation of protoporphyrinogen IX to the planar, fully conjugated macrocycle protoporphyrin IX in the inner membrane of the mitochondrion and requires oxygen for its activity.2 The PPOX activity is decreased to approximately half of the normal level in heterozygous VP patients.3 VP is inherited as an autosomal dominant trait displaying incomplete penetrance.4
The biochemical abnormalities found in VP patients include overproduction and increased excretion of porphyrins and porphyrin precursors. Faecal excretions of copro- and protoporphyrins are usually elevated together with urinary excretions of uro- and coproporphyrins. Plasma fluorescence spectrum shows an emission maximum at 626 nm, which is specific for VP.5,6,7,8 The sensitivity of these tests in symptom-free individuals is, however, less than 80%. Urinary porphobilinogen (PBG) and delta-aminolevulinic acid (ALA) are elevated during an acute attack and remain mildly elevated in remission in about 50% of patients.9
Clinical manifestations of VP include photosensitivity and acute neurovisceral attacks resembling other acute porphyrias. Photosensitivity manifests as skin fragility and blistering in sun-exposed areas. Excess porphyrins in plasma and/or skin interact with light energy inducing a phototoxic reaction and tissue damage.10 Symptoms of autonomic neuropathy include abdominal pain, vomiting, constipation, hypertension, and tachycardia.11,12 Peripheral neuropathies usually manifest as pain in the extremities or in the back and weakness that may progress to paresis.13 In the past, 17–38% of patients experienced acute attacks requiring hospitalisation,11,14,15 but milder symptoms of porphyria are more common occurring in 30–40% of patients.12 Acute attacks are often induced by precipitating factors such as drugs, alcohol, infection, fasting, or the menstrual cycle. The clinical onset of the disease usually occurs after puberty but probably more than 50% of the carriers of the affected gene remain symptom-free throughout their lives.11
The human PPOX-cDNA has been cloned from the human placental cDNA library16 and the PPOX gene mapped to chromosome 1q23.17,18 The gene is 5.5 kb in size including a 660 bp promoter region, and the coding region (1.5 kb) is spread over 13 exons.19 To date 111 mutations have been reported in the PPOX gene worldwide and no mutational hot spots have been identified.20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,4142 Thirty-eight (34%) of the mutations are small insertions or deletions, 44 (40%) are missense mutations, 17 (15%) change invariant nucleotides at splice sites, 1 (1%) is a gross deletion, and 11 (10%) produce stop codons.
In Finland to date, 143 VP patients, belonging to 21 families,11,12,41 have been biochemically and clinically well characterised. According to our data, the prevalence of VP is approximately 1.9 : 100 000 in the Finnish population of 5 000 000. Six of the Finnish VP mutations are family-specific and found so far only in Finland, whereas the major mutation (R152C), which was identified in 11 (52%) of the 21 Finnish VP families, has also been reported in France and in the USA.29,31 As previously reported,40,41 six of the mutations have been expressed in prokaryotic and eukaryotic cell systems. The PPOX activities of the mutated polypeptides were markedly reduced (⩽5%, Table 1) confirming that the mutations are responsible for the disease.
In this study, we have evaluated the clinical and biochemical outcome of 103 Finnish VP patients with mutations R152C, I12T, 338G→C, 78insC, IVS2-2a→c, 470A→C and 1203A→C. We have investigated the correlation between PPOX genotype and phenotype for the three most common Finnish mutations R152C, I12T and 338G→C. We have studied, (1) whether the occurrence of acute attacks or skin photosensitivity correlates with the mutation type, (2) whether the patients' biochemical characteristics differ depending on the mutation type, and (3) whether the occurrence of symptoms can be predicted by mutation type and/or biochemical tests in remission.
Subjects and methods
Patients and biochemical analyses
Since 1966, we have conducted a systematic follow-up of all Finnish patients known to have VP and informed them of the precipitating factors. For 14 of 21 VP families, ancestors could be traced back to the 18th or 19th centuries using church registers.11 Of the 143 VP patients diagnosed to date, 31 were deceased before 1966 and did not participate in the follow-up. Seven of them had died of an acute attack and an additional four had experienced acute attacks.12 In addition, four subjects under 14 years of age and one homozygous patient were excluded. Four subjects could not be traced for this study.
The diagnosis of VP was based either on mutation analysis (n=60), characteristic clinical symptoms with elevated faecal protoporphyrin excretion (n=68),43 typical plasma fluorescence emission spectrum (n=14),44 low lymphocyte PPOX activity in each family (n=25)3 and/or pedigree analysis (n=6). The mean PPOX activity measured from the patients' lymphocytes was 2.8±1.0 SD nmol/h/mg protein (range 1.1–6.2, normal 3.9–6.0, n=28).
PBG and ALA were measured using ALA/PBG Column test (Bio-Rad, CA, USA) based on Mauzerall and Granick.45 Until 1988, urinary excretions of uro- and coproporphyrin and faecal excretions of copro- and protoporphyrin were performed according to Rimington46 and Holti et al.47 Since 1988 all measurements were performed using high-pressure liquid chromatography (HPLC).43,48 The mean urinary copro- and uroporphyrin values and faecal copro- and protoporphyrin values of patients did not differ systematically when the method of measurement was changed. All biochemical measurements were performed in adolescence or adulthood (14 to 83 years). Porphyrins and porphyrin precursors were measured during remission of acute symptoms but in the presence, or absence, of skin disease.
Biochemical data were not obtained from 20 subjects (four symptomatic and 16 asymptomatic), of whom 14 were only screened with mutation analysis. Information about acute attacks was obtained from hospital records and personal interviews for all patients who had had acute attacks requiring hospitalisation between 1929 and 1966. Since 1966 the criteria for an acute attack were the acute nature of symptoms, urinary excretion of PBG at least five times above the upper limit of normal, and severe abdominal or other pain associated with one or more additional typical porphyric symptoms.11,12 The patients were examined clinically or interviewed to evaluate skin fragility in sun-exposed areas.
For mutation analysis (Table 1) DNA was isolated from blood leukocytes, amplified using polymerase chain reaction (PCR) -technique and either analysed using restriction digestion, whenever a specific enzyme was available, or by direct sequencing. The analyses were repeated at least twice for each sample in the presence of negative and positive controls.8 Informed consent was obtained for all DNA testing, and the study protocol was approved by the Ethical Committee of the Department of Medicine, University Central Hospital of Helsinki.
Statistical methods
Fischer's exact test was used for the comparison of categorical variables. Continuous variables were analysed using Mann–Whitney U-test, when two groups were compared, or Kruskal–Wallis one-way ANOVA, when more than two groups were compared simultaneously. Logistic regression with maximum likelihood estimation as optimisation criteria was employed to evaluate the association between dichotomous outcome variables (eg occurrence of skin symptoms or acute symptoms) and covariates (eg mutation group and biochemical tests). Statistical calculations were performed with SPSS version 10.04 and NCSS 2000.
Results
The study group consisted of 103 VP patients (36 male, 67 female, age 14–79 years), of whom 27 had experienced acute attacks, 41 photosensitivity and 14 both symptoms. Forty-nine were symptom-free throughout the follow-up period from 1966 to 2001. Twelve patients died during the follow-up. Fifty-two per cent of the patients experienced clinical manifestations before or during the follow-up period (Table 2 ). The overall frequency for skin symptoms was 40%, and for acute attacks 27%, respectively. The proportion of patients with acute symptoms had decreased dramatically from 38 to 14% among individuals diagnosed before and after 1980, whereas the prevalence of skin symptoms had decreased only subtly from 45 to 34% (Table 3 ). The decrease in acute symptoms was even more prominent among male patients, since none of the 17 patients diagnosed after 1980 had experienced acute attacks, whereas six of the 20 patients diagnosed before 1980 were symptomatic (P=0.02). The median age of the patients at the onset of acute symptoms was 30 years (range 17–55 years) and for skin symptoms 26 years (range 14–56 years), respectively. After the age of 40, only two patients experienced their first acute attacks and only one patient experienced her first skin symptoms.
Correlation between clinical symptoms and mutation type
The age of the patients at the time of diagnosis and gender distribution differed subtly among mutation groups (median age 30, 47 and 41 years and percentage of female patients 57, 75 and 73% for R152C, I12T and 338G→C, respectively). Of note is that none of the patients with I12T substitution manifested photosensitivity, and only one of 12 patients had experienced two acute attacks in her youth. The family became aware of VP mainly due to a homozygous patient with a severe phenotype.40 The occurrence of photosensitivity was significantly lower in the I12T group compared to the R152C group (P=0.001), whereas no significant differences between the R152C and 338G→C groups could be observed.
Correlation between the biochemical characteristics of patients and mutation type
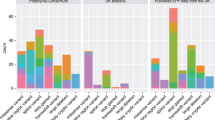
Figure 1 shows the biochemical data of the individuals with the three most common Finnish mutations. Overall, the urinary PBG and ALA were elevated in 73 and 52% of individuals with VP (data not shown). Urinary copro- and uroporphyrin were elevated in 59 and 72% and faecal coproporphyrin and protoporphyrin in 56 and 84% of the subjects, respectively. No significant difference existed between the excretions of the male and female patients.

Urinary and faecal porphyrin excretions of VP patients with the three most common mutations (n=69). The individual values were calculated as the mean of 1 to 7 single measurements in adulthood. Dashed line denotes the normal value limit and dash-dotted line shows the boundary between moderately and substantially elevated excretion.
The urinary excretions of uro- and coproporphyrins were significantly lower in the I12T group compared to the R152C group (P=0.001 for coproporphyrin and P=0.01 for uroporphyrin) and 338G→C group (P=0.01 for coproporphyrin). In addition, the excretions of PBG and ALA were at the lowest in the I12T group, although the differences between the groups were not significant (data not shown). The urinary excretions of uro- and coproporphyrins in the R152C and 338G→C groups were comparable. The faecal excretions of coproporphyrins and protoporphyrins were significantly lower in the I12T group compared to the R152C group (P=0.001 for coproporphyrin and P=0.0002 for protoporphyrin). Faecal excretion of protoporphyrin was also significantly lower in the I12T group than in the 338G→C group (P=0.01). The plasma fluorescence spectrum was normal in three patients tested in the I12T group, whereas in the R152C group it was positive in eight of nine patients and in the 338G→C group in two of three patients tested.
Correlation between biochemical characteristics and patients' symptoms
Table 4 shows the correlation between biochemical characteristics and patients' symptoms. The patients were defined in remission, if they had not experienced acute symptoms in 3 months, but may have manifested skin symptoms such as increased fragility of sun-exposed skin. The excretions of the patients, who are known to have experienced acute symptoms and/or skin symptoms, were compared to asymptomatic patients, who have never been clinically active. Urinary and faecal excretions of porphyrins were significantly higher in patients with prior symptoms, even during remission. This finding applied to all mutation groups. Figure 2D demonstrates that the faecal protoporphyrin test was 92% accurate in predicting those individuals who will remain symptom-free with respect to acute attacks. One patient with previous cyclical acute attacks, showed normal faecal protoporphyrin excretion in her post-menopausal phase. For skin symptoms, the negative predictive value was 92%, respectively. A 64-year-old patient, who had experienced mild skin fragility in his youth, had normal porphyrin excretion in faeces and urine, but a positive plasma fluorescence spectrum for VP. In two patients, a single measurement conducted before 18 years of age showed normal excretion even though the patients had skin symptoms and increased faecal protoporphyrin excretion later in adulthood. The negative predictive values of other biochemical tests varied from 61 to 77% for skin symptoms and 70 to 82% for acute attacks, when the normal value limits shown in Table 4 were used. In addition, urinary coproporphyrin excretion of more than 1000 nmol/day was associated with an increased occurrence of both skin symptoms and acute attacks with a positive predictive value of 100% (Figure 2A).

Clinical manifestations and porphyrin excretions of VP patients with seven different and two unknown mutations in remission (n=80). The values for individual persons were calculated as a mean of 1 to 7 single measurements in adulthood. Dashed line denotes the normal value limit.
Prediction of clinical symptoms
To predict the risk of clinical symptoms in previously asymptomatic patients, we constructed a logistic regression model49 where the occurrence of clinical symptoms was explained by the mutation type and/or biochemical tests. Because all patients with normal faecal protoporphyrin excretion were symptom-free at present, we included in the analysis only those individuals who had elevated excretion of fecal protoporphyrin. Table 5 shows that a moderate increase in urinary excretion of coproporphyrins in remission did not substantially increase the likelihood of symptoms, whereas a higher excretion (>1000 nmol, corresponding to the 80% percentage point of patients studied, normal value <236 nmol) was related to a significantly increased likelihood of skin symptoms, which was further increased by a markedly elevated excretion of protoporphyrin in the faeces (>644 nmol, normal value <130 nmol).
For an individual with normal faecal protoporphyrin excretion, the probability of developing skin symptoms is virtually zero. For the patients with normal urinary excretion of coproporphyrin and moderately elevated excretion of protoporphyrin in faeces, the estimated frequency of skin symptoms is 31%, whereas for an individual with a markedly increased excretion of urinary coproporphyrin and faecal protoporphyrin, the probability reaches 80% and the probabilities for other patients remain between these two extremes. A model, which could significantly predict the occurrence of acute attacks for these patients, could not be constructed.
Discussion
A systematic follow-up of Finnish VP patients, which was conducted from the early 1960s and based on hospital records from the beginning of the 20th century, provided an excellent opportunity for the genotype–phenotype analysis in VP. Our series includes both patients with symptoms (52%) and phenotypically normal carriers (48%) and thus provides information about the clinical and biochemical outcome among VP patients in general. The proportion of patients with acute attacks (27%) in this series is somewhat lower than in some extensive family studies in which up to 38% of patients had experienced acute attacks,14,31 but higher than 4–15% reported recently from a large South African kindred (Hift R, personal communication). The frequency of photosensitivity (40%) is lower than reported previously in South Africa and France (70%),14,31 but comparable with that of the recent South African study (39%) (Hift R, personal communication). Extended mutation screening among symptom-free family members and improved counselling explain the difference in these numbers.
The patients with the I12T mutation experienced no photosensitivity and acute attacks were rare. In addition, biochemical abnormalities were milder suggesting a milder form of the disease than in patients with other mutations. However, this could not be predicted by expression studies. The I12T mutation lies in the highly conserved FAD-binding domain in the amino-terminal region of the PPOX gene,16,50 whereas the R152C mutation causes an amino acid substitution in exon 5 and the 338G→C mutation results in exon 4 deletion and a truncated polypeptide. In vitro, the I12T mutation caused a dramatic decrease in the enzyme activity that was comparable to the activities found in the R152C and 338G→C mutations. Post-translational factors and interaction between the mutant and normal polypeptide may vary and modify the enzyme activity in vivo explaining the milder phenotype in patients with the I12T mutation. Interestingly, the homozygous patient with the I12T mutation demonstrated as high as 10% residual activity in vivo, which is sufficient to enable the patient to survive.40 Alternatively, another familial factor may be present in the subjects with the I12T mutation that influences their phenotype.
Urinary and faecal excretions of porphyrins in remission differed significantly between the mutation groups. Environmental influences may be important and other metabolic gene(s) that are presently not identified may modify the porphyrin excretion in general. Several polymorphisms in the cytochrome P450 enzymes exist51 and these genes are good candidates in searching genes modifying porphyrin metabolism and VP phenotype. Diminished supply of heme may lead to insufficient function of P450-mediated reactions in VP patients.52,53,54 P450 enzymes may also associate with the excretion of porphyrins, since CYP3A induction attenuated the hepatic accumulation and urinary excretion of uro- and heptacarboxylporphyrins in a rat porphyria cutanea tarda model.55
Since only 26% of symptomatic patients suffered both photosensitivity and acute attacks, the majority of the patients with each of these manifestations were in two distinct groups. This is in line with previous studies, where 79 and 77% of symptomatic patients experienced either photosensitivity or acute attacks, but not both.14,31 We have shown that the occurrence of acute attacks has decreased markedly during the last two decades, whereas no such tendency has been observed for skin symptoms.12,56 The decrease has been more prominent in males, since they are not prone to hormonal factors. This indicates that different pathogenetic mechanisms may underlie the development of skin symptoms and acute attacks. The latter may be more readily prevented by avoiding precipitating factors.
The occurrence of skin symptoms was related to a more than fourfold increase in urinary copro- and uroporphyrin excretion. In contrast, normal faecal protoporphyrin excretion as well as negative plasma fluorescence predicted freedom from skin symptoms. These findings support the theory that the severity of chronic skin symptoms is likely to depend on the permanent circulating levels of porphyrins.10,56 In the pathogenesis of acute attacks, the induction of ALA-synthase (ALAS), which is the rate-limiting enzyme of the pathway, plays a key role. Moreover, if heme requirements are increased in the liver, ALAS is induced resulting in the accumulation of porphyrins and porphyrin precursors in those patients.57
In our series, normal excretion of protoporphyrin in faeces in adulthood predicted freedom from both skin symptoms and acute attacks for patients. In contrast, normal urinary excretion of porphyrins, PBG or ALA, or normal faecal excretion of coproporphyrin did not predict freedom from symptoms for VP patients. The most valuable test predicting an increased risk of symptoms was urinary coproporphyrin, but only a substantially increased excretion exceeding 1000 nmol/day was associated with an increased risk of both skin symptoms and acute attacks and virtually all patients with an excretion of more than 1000 nmol/day experienced either skin symptoms, acute attacks, or both.
Electronic-Database information
Human Gene Mutation Database, http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html (for mutations in the PPOX gene); Human Genome Mapping Project, http://www.hgmp.mrc.ac.uk (for accession numbers for PPOX genomic/cDNA sequences: X99450 [human]); Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for VP [MIM 176200]).
References
Brenner DA, Bloomer JR . The enzymatic defect in variegate porphyria N Engl J Med 1980 302: 765–769
Dailey HA . Conversion of coproporphyrinogen to proto hem in higher eukaryotes and bacteria: terminal three enzymes in Dailey HA (ed) Biosynthesis of heme and chlorophylls New York: McGraw-Hill 1990 pp 123–161
Deybach JC, de Verneuil H, Nordmann Y . The inherited enzymatic defect in porphyria variegata Hum Genet 1981 58: 425–428
Kappas A, Sassa S, Galbraith RA, Nordmann Y . The porphyrias in Scriver CR, Beaudet A, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited diseases New York: McGraw-Hill 1995 pp 2116–2127
Long C, Smyth SJ, Woolf J et al. Detection of latent variegate porphyria by fluorescence emission spectroscopy of plasma Br J Dermatol 1993 129: 9–13
Enriquez de Salamanca R, Sepulveda P, Moran MJ, Santos JL, Fontanellas A, Hernandez A . Clinical utility of fluorometric scanning of plasma porphyrins for the diagnosis and typing of porphyrias Clin Exper Dermatol 1993 18: 128–130
Da Silva V, Simonin S, Deybach JC, Puy H, Nordmann Y . Variegate porphyria: diagnostic value of fluorometric scanning of plasma porphyrins Clin Chim Acta 1995 238: 163–168
von und zu Fraunberg M, Kauppinen R . The diagnosis of variegate porphyria – Hard to Get? Scand J Clin Lab Invest 2000 60: 1–6
Meissner PN, Adams P, Kirsch R . Allosteric inhibition of human lymphoblasts and purified porphobilinogen deaminase by protoporphyrinogen and coproporphyrinogen: a possible mechanism for the acute attack of variegate porphyria J Clin Invest 1993 91: 1436–1444
Day RS . Variegate porphyria Semin Dermatol 1986 5: 138–154
Mustajoki P . Variegate porphyria. Twelve years' experience in Finland Q J Med 1980 49: 191–203
Kauppinen R, Mustajoki P . Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases Medicine (Baltimore) 1992 71: 1–13
Ridley A . The neuropathy of acute intermittent porphyria Q J Med 1969 38: 307–333
Eales L, Day RS, Blekkenhorst GH . The clinical and biochemical features of variegate porphyria: an analysis of 300 cases studied at Groote Schuur Hospital, Cape Town Int J Biochem 1980 12: 837–853
Muhlbauer JE, Pathak MA, Tishler PV et al. Variegate porphyria in New England JAMA 1982 247: 3095–3102
Nishimura K, Taketani S, Inokuchi H . Cloning of a human cDNA for protoporphyrinogen oxidase by complementation in vivo of a hemG mutant of Escherichia coli J Biol Chem 1995 270: 8076–8080
Taketani S, Inazawa J, Abe T et al. The human protoporphyrinogen oxidase gene (PPOX): organization and location to chromosome 1 Genomics 1995 29: 698–703
Roberts AG, Whatley SD, Daniels J et al. Partial characterization and assignment of the gene for protoporphyrinogen oxidase and variegate porphyria to human chromosome 1q23 Hum Mol Genet 1995 4: 2387–2390
Puy H, Robreau AM, Rosipal R, Nordmann Y, Deybach JC . Protoporphyrinogen oxidase: complete genomic sequence and polymorphisms in the human gene Biochem Biophys Res Commun 1996 226: 226–230
Meissner PN, Dailey TA, Hift RJ et al. A R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria [see comments] Nat Genet 1996 13: 95–97
Warnich L, Kotze MJ, Groenewald IM et al. Identification of three mutations and associated haplotypes in the protoporphyrinogen oxidase gene in South African families with variegate porphyria Hum Mol Genet 1996 5: 981–984
Deybach JC, Puy H, Robreau AM et al. Mutations in the protoporphyrinogen oxidase gene in patients with variegate porphyria Hum Mol Genet 1996 5: 407–410
Lam H, Dragan L, Tsou HC et al. Molecular basis of variegate porphyria: a de novo insertion mutation in the protoporphyrinogen oxidase gene Hum Genet 1997 99: 126–129
Frank J, Christiano A . Genetic research strategies: a review of the acute porphyrias Retinoids 1997 13: 88–92
Frank J, Jugert FK, Kalka K, Goerz G, Merk HF, Christiano AM . Variegate porphyria: identification of a nonsense mutation in the protoporphyrinogen oxidase gene J Invest Dermatol 1998 110: 449–451
Frank J, McGrath J, Lam H, Graham RM, Hawk JL, Christiano AM . Homozygous variegate porphyria: identification of mutations on both alleles of the protoporphyrinogen oxidase gene in a severely affected proband J Invest Dermatol 1998 110: 452–455
Frank J, Lam H, Zaider E, Poh-Fitzpatrick M, Christiano AM . Molecular basis of variegate porphyria: a missense mutation in the protoporphyrinogen oxidase gene J Med Genet 1998 35: 244–247
Roberts AG, Puy H, Dailey TA et al. Molecular characterization of homozygous variegate porphyria Hum Mol Genet 1998 7: 1921–1925
Frank J, Poh-Fitzpatrick MB, King Jr LE, Christiano AM . The genetic basis of ‘Scarsdale Gourmet Diet’ variegate porphyria: a missense mutation in the protoporphyrinogen oxidase gene Arch Dermatol Res 1998 290: 441–445
Corrigall AV, Hift RJ, Hancock V et al. Identification and characterisation of a deletion (537delAT) in the protoporphyrinogen oxidase gene in a South African variegate porphyria family Hum Mutat 1998 12: 403–407
Whatley SD, Puy H, Morgan RR et al. Variegate porphyria in Western Europe. Identification of PPOX gene mutations in 104 families, extent of allelic heterogenity, and absence of correlation between phenotype and type of mutation Am J Hum Genet 1999 65: 984–994
De Siervi A, Parera VE, del C Batlle AM, Rossetti MV . Two new mutations (H106P and L178V) in the protoporphyrinogen oxidase gene in Argentinean patients with variegate porphyria Hum Mutat 2000 16: 532
De Siervi AD, Parera VE, Varela LS, del C Batlle AM, Rossetti MV . A novel mutation (1320InsT) identified in two Argentine families with variegate porphyria Hum Mutat 2000 16: 96
Maeda N, Horie Y, Sasaki Y et al. Three novel mutations in the protoporphyrinogen oxidase gene in Japanese patients with variegate porphyria Clin Biochem 2000 33: 495–500
Frank J, Aita VM, Ahmad W, Lam H, Wolff C, Christiano AM . Identification of a founder mutation in the protoporphyrinogen oxidase gene in variegate porphyria patients from Chile Hum Hered 2001 51: 160–168
Frank J, Jugert JK, Merk HF et al. A spectrum of novel mutations in the protoporphyrinogen oxidase gene in 13 families with variegate porphyria J Invest Dermatol 2001 116: 821–823
Palmer RA, Elder GH, Barrett DF, Koehane SG . Homozygous variegate porphyria: a compound heterozygote with novel mutations in the protoporphyrinogen oxidase gene Br J Dermatol 2001 144: 866–869
Lam CW, Hui KN, Poon PM et al. Novel splicing mutation of the PPOX gene (IVS10 + 1G→A) detected by denaturing high-performance liquid chromatography Clin Chim Acta 2001 305: 197–200
Corrigall AV, Hift RJ, Davids LM et al. Identification of the first variegate porphyria mutation in an indigenous black South African and further evidence for heterogeneity in variegate porphyria Mol Genet Metabol 2001 73: 91–96
Kauppinen R, Timonen K, von und zu Fraunberg M et al. Homozygous variegate porphyria: 20 years' follow up and characterization of molecular defect J Invest Dermatol 2001 116: 610–613
von und zu Fraunberg M, Tenhunen R, Kauppinen R . Expression and characterization of six mutations in the protoporphyrinogen oxidase gene among Finnish variegate porphyria patients Mol Med 2001 7: 320–328
Corrigall AV, Hift RJ, Davids LM et al. Homozygous variegate porphyria in South Africa: genotypic analysis in two cases Mol Genet Metabol 2000 69: 323–330
Li F, Lim CK, Peters TJ . Analysis of urine and faecal porphyrins by HPLC coupled to an advanced automated sample processor Biomed Chromatogr 1986 1: 93–94
Poh-Fitzpatrick MB . A plasma porphyrin fluorescence marker for variegate porphyria Arch Dermatol 1980 116: 543–547
Mauzerall D, Granick S . The occurrence and determination of delta-aminolevulinic acid and porphobilinogen in urine J Biol Chem 1956 219: 435–436
Rimington C . Quantitative determination of porphobilinogen and porphyrin in urine and faeces Association of Clinical Pathology 1958 Broadsheet No. 21
Holti R, Rimington C, Tate BC, Thomas C . An investigation of porphyria cutanea tarda Q J Med 1958 27: 1–17
Lim CK, Peters TJ . Urine and faecal porphyrin profiles by reversed-phase high-performance liquid chromatography in the porphyrias Clin Chim Acta 1984 139: 55–63
Hosmer DW, Lemeshow S . Applied Logistic Regression New York: John Wiley & Sons 1989
Dailey TA, Dailey HA . Human protoporphyrinogen oxidase: Expression, purification, and characterization of the cloned enzyme Protein Sci 1996 5: 98–105
van der Weide J, Steijns LS . Cytochrome P450 enzyme system: genetic polymorphisms and impact on clinical pharmacology Ann Clin Biochem 1999 36: 722–729
Birnie GG, McColl KEL, Thompson GG, Moore MR, Goldberg A, Brodie MJ . Antipyrine metabolism in acute hepatic porphyria in relapse and remission Br J Clin Pharmacol 1987 23: 358–361
Tokola O, Mustajoki P, Himberg JJ . Haem arginate improves hepatic oxidative metabolism in variegate porphyria Br J Clin Pharmacol 1988 26: 753–757
Mustajoki P, Mustajoki S, Rautio A, Arvela P, Pelkonen O . Effects of heme arginate on cytochrome P450-mediated metabolism of drugs in patients with variegate porphyria and healthy men Clin Pharmacol Ther 1994 56: 9–13
Franklin MR, Phillips JD, Kushner JP . CYP3A-inducing agents and the attenuation of uroporphyrin accumulation and excretion in a rat model of porphyria cutanea tarda Biochem Pharmacol 2000 60: 1325–1331
Timonen K, Niemi KM, Mustajoki P, Tenhunen R . Skin changes in variegate porphyria. Clinical, histopathological, and ultrastructural study Arch Dermatol Res 1990 282: 108–114
Meyer UA, Schuurmans MM, Lindberg RLP . Acute porphyrias: pathogenesis of neurological manifestations Semin Liver Dis 1998 18: 43–52
Acknowledgements
This study was supported by grants from the Magnus Ehrnrooth Foundation, the Instrumentarium Research Foundation, Jalmari and Rauha Ahokas Foundation, the Research Funds and the Clinical Research Institute of the Helsinki University Central Hospital, the Biomedicum Helsinki Foundation and the University of Helsinki.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
von und zu Fraunberg, M., Timonen, K., Mustajoki, P. et al. Clinical and biochemical characteristics and genotype–phenotype correlation in Finnishvariegate porphyria patients. Eur J Hum Genet 10, 649–657 (2002). https://doi.org/10.1038/sj.ejhg.5200860
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200860
Keywords
This article is cited by
-
Partial protoporphyrinogen oxidase (PPOX) gene deletions, due to different Alu-mediated mechanisms, identified by MLPA analysis in patients with variegate porphyria
Orphanet Journal of Rare Diseases (2013)
-
The incidence of inherited porphyrias in Europe
Journal of Inherited Metabolic Disease (2013)
-
Clinic and genetic evaluation of variegate porphyria (VP) in a large family from the Balearic Islands
Journal of Inherited Metabolic Disease (2009)
-
Genetic and biochemical studies in Argentinean patients with variegate porphyria
BMC Medical Genetics (2008)
