Article Text

Abstract
Background and aims: The incidence of complement abnormalities in the UK is not known. It is suggested in at least three major paediatric textbooks to test for abnormalities of the complement system following meningococcal disease (MCD).
Methods: Over a four year period, surviving children with a diagnosis of MCD had complement activity assessed. A total of 297 children, aged 2 months to 16 years were screened.
Results: All children except one had disease caused by B or C serogroups. One child, with group B meningococcal septicaemia (complicated by disseminated intravascular coagulation and who required ventilation and inotropic support) was complement deficient. C2 deficiency was subsequently diagnosed. She had other major pointers towards an immunological abnormality prior to her MCD.
Conclusion: It is unnecessary to screen all children routinely following MCD if caused by group B or C infection. However, it is important to assess the previous health of the child and to investigate appropriately if there have been previous suspicious infections, abnormal course of infective illnesses, or if this is a repeated episode of neisserial infection.
- meningococcal disease
- complement deficiency
- MCD, meningococcal disease
- PCR, polymerase chain reaction
Statistics from Altmetric.com
Meningococcal disease (MCD) may be associated with abnormalities of the complement system. Several authoritative paediatric texts recommend screening for complement deficiencies following MCD,1–3 but there have been no large UK paediatric studies to establish whether this is worthwhile. These disorders, if identified, have implications for management, including family studies, prophylaxis, vaccination, and altered thresholds for infection screening and treatment. However, testing is time consuming (especially when families need to travel long distances to clinics), needs to be done several weeks after recovery, and requires specialist laboratory services and interpretation. Should all UK children with MCD therefore be screened for complement deficiencies, or is a selective policy more appropriate? We measured complement activity following recovery from MCD in an unselected series of children, in order to estimate the incidence of undiagnosed complement deficiencies and to identify factors which might warn of this diagnosis.
SUBJECTS AND METHODS
From 1996 to 1999, children with a provisional diagnosis of MCD were identified. These were patients seen at, referred to, or discussed with either the Regional Paediatric Infectious Diseases Unit or the Paediatric Intensive Care Unit Retrieval Service, from units in the North East Region. Surviving children with suspected or proven MCD were invited for review in the clinic 6–8 weeks following discharge. Providing there were no ongoing inflammatory processes (such as necrotic skin lesions, new/different infections, or recent surgery) CH100 and AP100 were measured in those children with proven MCD (based on culture and non-culture methods) and in those where a clinical diagnosis had been made. These are gel assays and measure the integrity of the classical and alternative pathways of complement activation.4 If CH100 or AP100 were abnormal, additional tests were done to identify the missing complement components.
RESULTS
A total of 824 children (age range 2 months to 16 years), with a provisional diagnosis of MCD were identified; 60% were under 5 years of age. Four had a previous medical history of note: two with pneumonia, and one with urinary tract infection with vesicoureteric reflux. The fourth case is discussed subsequently. There was no relevant family history of problems potentially related to complement abnormalities in any child.
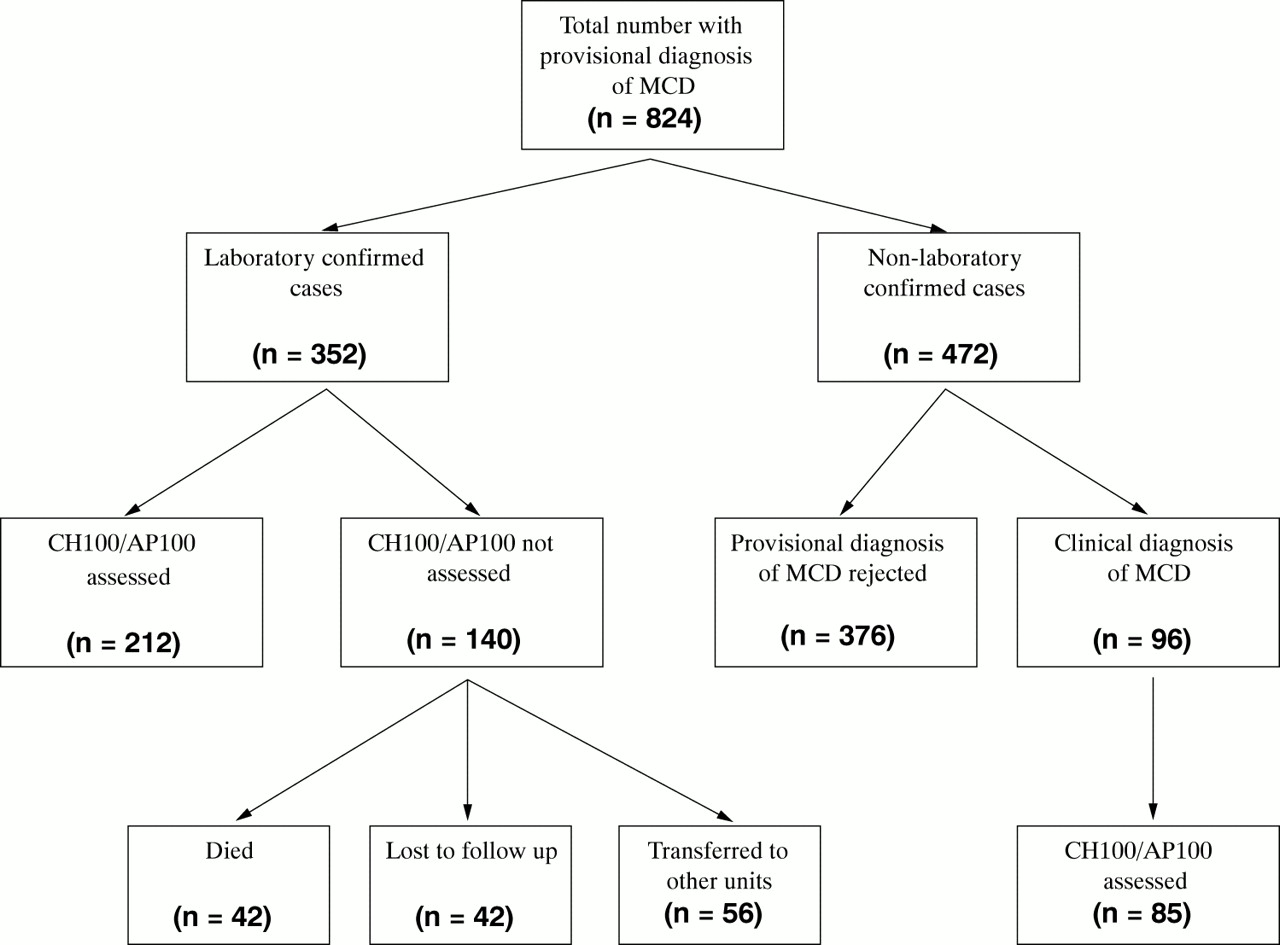
A total of 118/824 (14%) required intensive care; there were 42 deaths (5%). In 352 there was laboratory confirmation (cultures, polymerase chain reaction (PCR), or serology) of infection with Neisseria meningitidis. Complement assays were performed in 212; assays were not done in 140 because of death, non-attendance, or discharge back to distant units (fig 1). Group B accounted for 203 cases, group C 138 cases, 11 were caused by non-groupable N meningitidis, and one case was caused by W135.

{kind=link}
Breakdown of numbers of children with a provisional diagnosis of MCD who were then screened for abnormalities of the complement system. Only one of these children was complement deficient.
Of the remaining 472 unconfirmed cases, 376 (who mainly presented with a petechial rash but without severe sepsis or shock) were treated with antibiotics for 48–72 hours which were then stopped on obtaining negative cultures. Ninety six, although culture and/or PCR negative, had a clinical diagnosis of MCD and were treated accordingly; 85 of this group had gel assays performed at follow up. Of the total of 297 complement assays done, there was only one child with an abnormal result (see below).
Case report
This 4 year old child presented with a 24 hour history of high fever, vomiting, and a petechial rash. On admission she was shocked and required immediate fluid resuscitation. A total of 65 ml/kg of colloid was required within the first 24 hours. She was intubated, ventilated, and required inotropic support with dobutamine (maximum dose 12 μg/kg/min), adrenaline (maximum dose 2 μg/kg/min), and subsequently noradrenaline (0.5 μg/kg/min). High dose intravenous cefotaxime was commenced (200 mg/kg/day). She developed a disseminated intravascular coagulopathy (prothrombin time 27 sec, activated partial thromboplastin time 59.9 sec, fibrinogen 1.6 g/l, d-dimers 4000 ng/ml) and was treated with fresh frozen plasma and cryoprecipitate. She was ventilated for five days; her course was complicated by extensive oral infection with Herpes simplex (requiring intravenous acyclovir), an adenoviral chest infection, staphylococcal skin sepsis, a left knee effusion, and necrotic skin areas related to her purpuric rash. Neisseria meningitidis (group B) was cultured both from blood and from a petechial lesion; she also had a positive PCR for blood meningococcus.
Previously, at the age of 4 months, she had an illness characterised by high temperature, irritability, and then a focal fit. On admission to her local hospital at that time, she was shocked and had required fluid resuscitation, high dose benzylpenicillin, and cefotaxime. A septic screen revealed Pneumococcus in both blood and cerebrospinal fluid. She made a successful but slow recovery from this illness, though she also developed respiratory syncytial virus positive bronchiolitis during convalescence. There was no relevant family history of note and in particular no history of meningitis, recurrent infections, or unexplained deaths in infancy. Her five siblings were all in good health as were her non-consanguineous parents.
After recovering from MCD, she was allowed home and was reviewed two months later in the clinic when CH100 and AP100 were measured on a fresh sample, sent directly to the laboratory. AP100 was normal, but CH100 activity was completely absent. C2 deficiency was subsequently confirmed by a double diffusion technique, with no detectable C2 activity. There was no clinical or laboratory evidence of autoimmune disease and autoantibodies have been negative. Family studies revealed no other affected members and other immunological tests (full blood count, immunoglobulins and IgG subclasses, IgM isohaemagglutinins, antibody responses to diphtheria, tetanus, Haemophilus influenzae type B and Pneumococcus vaccines, lymphocyte subsets, C3 and C4, and abdominal ultrasound scan) were all normal.
She has been followed up over the past two years, although her attendance has been poor, and has had no further significant infections. Antibiotic prophylaxis with co-trimoxazole has been prescribed, and she has been immunised with pneumococcal and tetravalent meningococcal polysaccharide vaccines.
DISCUSSION
The complement system is a series of 19 plasma and nine membrane proteins, first discovered at the turn of the twentieth century. However it was not until the 1960s that the first complement deficient patient was described.3 The system is usually divided into classical (where antibody–antigen interactions initiate the complement cascade) and alternative pathways (which are independent of such) and are described elsewhere.2, 3 There are distinct geographical and ethnic influences on the incidence of complement deficiencies.2, 5 Thus in Japan, C9 deficiency may occur in up to 0.1% of the population, although it is uncommon in white people. Conversely C2 deficiency has a frequency of 0.01% in the USA, although it is unreported in Japan.5 Complement deficiency was found in 6/176 Dutch patients (3%) over a 33 year period.6 Thus the threshold for screening for complement abnormalities should reflect these epidemiological variations.
Primary complement deficiencies are closely associated with increased susceptibility to infection, rheumatic/autoimmune diseases, or angioedema. Properdin deficiency is X linked. Deficiencies in either the classical or alternative pathways which lead to suboptimal cleavage/activation of C3 (an important opsonic ligand) render children more susceptible to encapsulated bacterial infections, as opsonisation is the primary host defence against these organisms. Deficiencies in the terminal components C5–C9 lead to poor bactericidal function, as these are the components needed to form the membrane attack complex. While Gram positive infections are effectively dealt with in these late component deficiencies (as opsonisation is normal), Gram negative infections, most notably with Neisseria spp (which rely on serum bactericidal activity), are not. Late component deficient patients in the US population may have a 1000–10 000-fold increased risk of infection, with a median age of onset in the mid-teens (compared to the first five years in the normal population), a relapse rate of approximately 8%, and a recurrence rate of 44%.5 Thus repeated or unusual infections caused by Neisseria spp should alert clinicians to the possibility of both terminal complement abnormalities as well as properdin deficiency, a component of the alternative pathway.7
It has also been postulated that infection with unusual serogroups (such as W135 and Y in the UK), or with non-groupable N meningitidis should prompt investigation of the patient's immune system,7–9 perhaps related to the propensity of unusual serogroups to cause disease in older individuals who have an increased lifelong susceptibility. Somewhat paradoxically, mortality rates in meningococcal infection in complement deficient patients are better than those for the normal population (1.5 v 19%); Figueroa et al explore the possible reasons for this. The fact that these patients are complement deficient may work to their advantage: a decrease in endotoxin related complement activation may lead to less severe clinical disease, shock, cerebral oedema, coagulation defects, and tissue injury compared to complement sufficient patients.5
Correctly identifying children with complement abnormalities is important and worthwhile. This is consequential pre-emptive information for families and medical carers, and should alter thresholds for the early investigation of and intervention for possible infective episodes. In our unit it is felt mandatory to assess CH100/AP100 (going on to more specific assays as necessary) if any of the following factors are present:
Repeated neisserial infections
Infection with an unusual serogroup
Fulminant disease in males (properdin deficiency)
Other pointers towards immunodeficiency (such as repeated or unusual infections with other organisms, family history, unusual course of the illness, etc)
Coexisting angio-oedema, autoimmune, or connective tissue disorders.
If a complement deficiency is identified, antibiotic prophylaxis should be considered, and special attention should be given to immunisation against encapsulated organisms such as pneumococcus, Haemophilus, and Neisseria. The tetravalent polysaccharide vaccine for meningococcal groups A, C, Y, and W135 may be offered. In addition, group C meningococcus conjugate vaccine is also now freely available in the UK. Early identification and management of both autoimmune and allergic phenomena may also be feasible. The primary complement deficiencies are inherited as autosomal codominants; heterozygosity results in approximately half normal levels of the protein,2 and family studies may identify other affected members.It has been proposed in three major textbooks of paediatrics that all cases of meningococcal infection should have complement assays assessed1–3 (though in the experience of the authors this is not widely practised in the UK). This advice is based on studies describing incidence of deficiencies in 5–18% of cases after a single neisserial infection. Tedesco et al, in a collation of seven studies, suggested that 14% of patients with sporadic MCD might be expected to have a defect in C5–9,10 rising to one third (for any component) in those with more than one episode. Ellison et al described a series of 20 patients, all adults, all from North America, with a single episode of meningococcal meningitis, septicaemia, or pericarditis; six had complement abnormalities. However, although three had a deficiency of a terminal pathway component (C6 in two, C8 in one), the rest had deficiencies of multiple complement components associated with systemic lupus erythematosus or multiple myeloma.11 Thus these reports and recommendations may not be applicable to the general paediatric population in the UK with MCD.This large study of 297 children indicates that routine screening for complement deficiencies of all children in the UK following MCD is unlikely to be useful or cost effective. An individual assay for CH100 and AP100 costs approximately £25 per case (personal communication, Dr A Fay). In addition, the logistics of outpatient assessment, medical, nursing, and laboratory time, plus a potentially unnecessary venepuncture, also need to be taken into account. Over a four year period the only child identified with a complement deficiency had clear pointers to an immunological abnormality. It is also interesting that her abnormality was not in the terminal complement components, but C2 deficiency. This is the commonest of the inherited deficiencies, variable in clinical presentation, with increased susceptibility to infection (particularly blood borne, systemic sepsis caused by encapsulated bacteria) and a variety of rheumatic/autoimmune diseases (systemic lupus erythematosus, glomerulonephritis, dermatomyositis, etc).3A selective policy for post-MCD screening in the UK is suggested. It is important to assess the past medical history and to investigate appropriately if there have been previous suspicious infections, an abnormal course of infective illnesses, infection with non-groupable or unusual serogroups, or repeated episodes of neisserial infection. However, if no such factors are present, testing should not be obligatory.uthors' affiliations