Article Text

Abstract
The optimum approach to congenital leukaemia is unclear. Results of treatment are generally discouraging and palliation is offered to many. The successful treatment of an infant with congenital leukaemia is reported.
- congenital
- leukaemia
- chemotherapy
- bone marrow transplantation
- white blood cells
Statistics from Altmetric.com
Case report
A female infant weighing 2790 g was born at term by emergency caesarean section because of fetal distress. Soon after birth she needed resuscitation and ventilation. The only abnormal physical signs were pallor and massive hepatomegaly. Haemoglobin was 9.9 g/dl, white blood cell count 357 × 109/l, and platelet count 10 × 109/l, and 95% of cells were moderate sized blasts with high nuclear/cytoplasmic ratio and a single nucleolus but no Auer rods or granules. The blasts were negative for Sudan black, myeloperoxidase, chloroacetate, and non-specific esterase. They were negative for CD2, 3, 7, 10, 19, 61 and surface immunoglobulin, but positive for CD13 and CD33. Cytogenetic investigations showed 46X, ins(X;6)(p11;q1 q1), a balanced insertional translocation relocating a segment of proximal 6q into proximal Xp, but no evidence of bcr/abl or MLL gene rearrangements. Bone marrow examination showed more than 90% blasts, identical with those in the blood. The differential diagnoses comprised “null” acute lymphoblastic leukaemia (ALL) expressing myeloid markers, or M0 (minimally differentiated) acute myeloblastic leukaemia (AML).
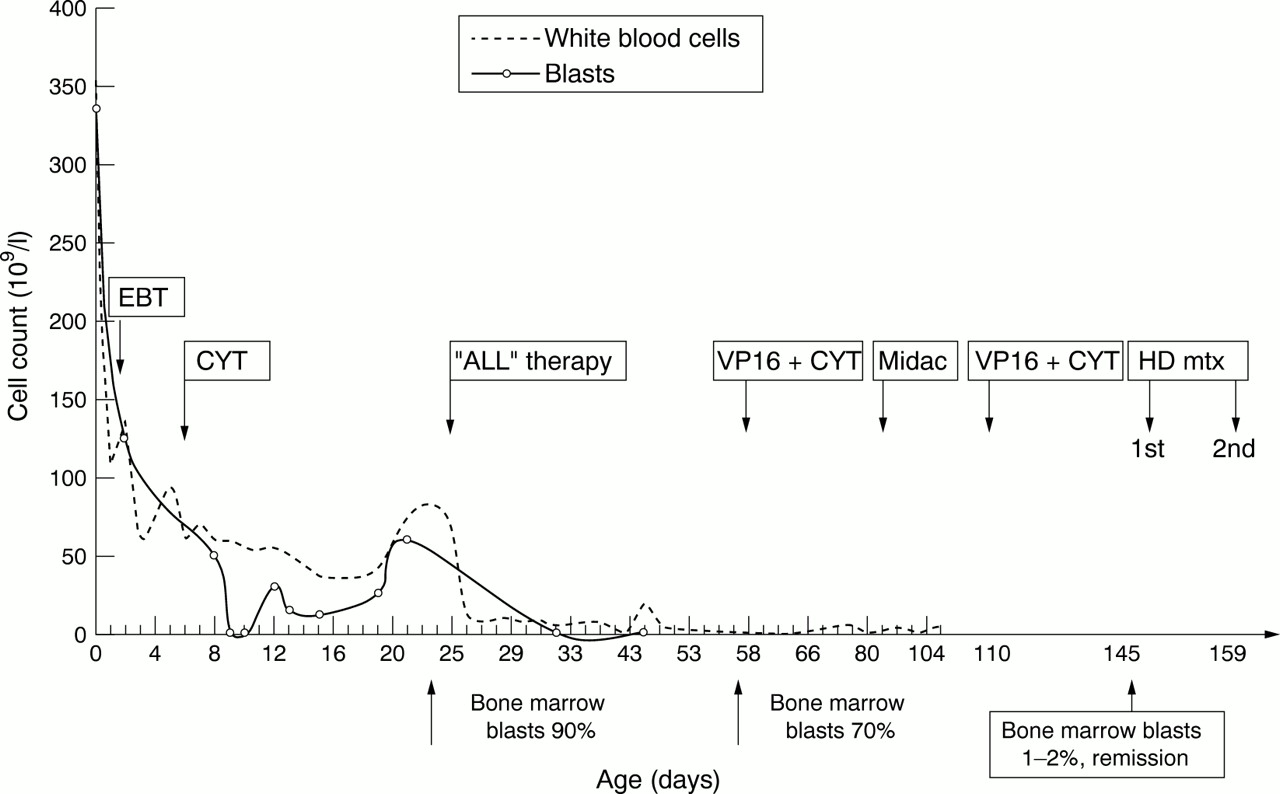
Figure 1 shows a summary of her management and response to treatment in the first few months. She had a double volume exchange transfusion on day 2 for leucostasis after which the white blood cell count fell to 100 × 109/l. Intravenous cytarabine 7.5 mg was given on day 4. During seven days of ventilatory support, she had a single generalised seizure; electroencephalogram and ultrasound of the head were normal. Subsequently, she was well enough to be transferred to the paediatric oncology unit on day 21. Repeat bone marrow assessment showed no change. Examination of the cerebral spinal fluid showed 0.06 × 109 cells/l; 30% were blasts, confirming central nervous system involvement. Formal chemotherapy began on day 23 with vincristine, prednisolone, and asparaginase. Although the peripheral blood blast cell count fell, after four weeks a repeat bone marrow assessment still showed 70% blasts. Nevertheless, as the infant's general condition had improved dramatically, it was decided to continue chemotherapy with a hybrid of ALL and AML regimens.
{kind=link}
Effect of chemotherapy on peripheral white blood count and blast count. EBT, Exchange blood transfusion; VP16, etoposide; CYT, cytarabine; ALL therapy, acute lymphoblastic leukaemia treatment; Midac, mitozantrone and cytarabine; HD mtx, high dose methotrexate.
The baby received courses of intravenous etoposide/cytarabine and mitozantrone/cytarabine (fig 1). This led to complete clinical and morphological remission by day 145 of age. Central nervous system directed treatment was given with intravenous high dose methotrexate and 11 doses of intrathecal methotrexate. Apart from the second course of high dose methotrexate, all chemotherapy doses were reduced by 50% of the standard calculated dose on the basis of her body surface area.
No HLA matched family donor was available, but an HLA-A, B, DRB1* matched unrelated donor was identified. Bone marrow transplantation was performed during the first complete remission at 7 months of age. Conditioning comprised oral busulphan (18 mg/kg) and cyclophosphamide (200 mg/kg). Prophylaxis against graft rejection was given using intravenous CAMPATH 1G, and graft versus host disease prophylaxis included use of in vitro T cell depletion of donor marrow (using CAMPATH 1M and complement) and cyclosporin A after transplantation. CAMPATH 1G and CAMPATH 1M are anti-lymphocyte antibodies supplied by the Therapeutic Antibody Centre (University of Oxford, Oxford, UK). The infant engrafted slowly, and received granulocyte colony stimulating factor, before achieving a neutrophil count of 0.5 × 109/l by day +33. She did not develop graft versus host disease. Hepatic veno-occlusive disease was diagnosed on day +18 (peak weight gain 22% and serum bilirubin 177 μmol/l), and was treated with intravenous recombinant tissue plasminogen activator and heparin.
Interstitial pneumonitis was diagnosed on day +48, with no evidence of infection, and was treated with intravenous and nebulised steroids. The infant recovered fully and was discharged home on day +90. She has remained well during follow up of 28 months since the bone marrow transplantation, with normal developmental progress and no evidence of leukaemic relapse. DNA analysis of peripheral blood cells has confirmed donor chimerism.
Discussion
The absence of specific cytochemical, immunophenotypic, and cytogenetic features prevented a more precise diagnosis than acute leukaemia. Because of the infant's poor general condition at birth, it was difficult to decide whether to treat her, and if so, with what. The response to cytarabine and subsequent improvement in her condition encouraged us to pursue a therapeutic rather than palliative approach. However, there is high morbidity and mortality following chemotherapy in congenital leukaemia. In a population based series of 15 patients, 13 died during primary management, 11 within 30 days of diagnosis (A R Craig, R Feltblower and S E Kinsey, Proceedings of the British Society of Haematology, 1999). As the blasts morphologically resembled lymphoblasts, we opted for a less toxic induction treatment using a typical ALL regimen. The partial response suggests that some of the blasts were of lymphoid origin. The baby's continued general improvement encouraged us to give her further treatment with a potentially more toxic regimen. The initial response to cytarabine led us to include this drug in her subsequent treatment.
In view of the relatively late achievement of complete remission, and the generally very poor outcome of infants treated with chemotherapy alone for congenital leukaemia,1 it was decided that allogeneic bone marrow transplantation should be performed during the first complete remission. Fortunately, a well matched unrelated donor was identified in the absence of a suitable related donor. The outcome of bone marrow transplantation in children using unrelated donors has improved greatly during the last decade and is now comparable with that of HLA-identical sibling donor transplantations in some series.2 Unrelated donor bone marrow transplantation is regarded as an acceptable treatment in first complete remission for certain subgroups of high risk childhood acute leukaemia—for example, Philadelphia chromosome positive ALL.3 However, there are very few case reports of the use of bone marrow transplantation in congenital leukaemia.1 The occurrence in our patient of veno-occlusive disease and pneumonitis, two potentially fatal complications after bone marrow transplantation, illustrates its hazardous nature. Nevertheless, in congenital acute leukaemia, which is rarely cured by chemotherapy alone, we believe that the use of bone marrow transplantation, using an unrelated donor if necessary, should at least be considered as a potential treatment.