Article Text

Abstract
Background and aims: Reticulo-endothelial macrophages together with duodenal enterocytes coordinate body iron homeostasis. The aim of this study was to investigate the regulatory actions of the hormone hepcidin on ferroportin expression in these two cell types.
Methods: We investigated the in vitro effects of hepcidin in well-characterised human cell culture models of macrophages (differentiated THP-1 cells) and intestinal epithelial cells (Caco-2 cells). The in vivo effects of hepcidin were also investigated in mice injected with a synthetic hepcidin peptide.
Results: Exposure to hepcidin (presented either as conditioned medium from interleukin-6-stimulated HuH7 cells or as a synthetic peptide) resulted in a rapid (within 4 h) decrease in ferroportin expression in THP-1 macrophages but had no effect on ferroportin levels in Caco-2 cells. To determine whether these rapid effects of hepcidin were also evident in vivo we injected mice with a synthetic hepcidin peptide. Four hours post-injection, ferroportin levels in the macrophage-rich red pulp of the spleen were decreased significantly and the hepcidin-treated mice developed hypoferraemia. Interestingly, in the same mice there was no effect of hepcidin on duodenal ferroportin protein expression or duodenal iron transport.
Conclusions: These data suggests that the rapid response to hepcidin is cell type and tissue specific. Upon its release, hepcidin initially targets macrophage iron recycling. The duodenum appears to be less sensitive to this initial rise in hepcidin levels. We believe the fact that macrophages respond more acutely to a hepcidin challenge is fully consistent with their central role in maintaining body iron homeostasis.
Statistics from Altmetric.com
Body iron homeostasis is maintained through the absorption of 1–2 mg/day of iron from the diet by the duodenal enterocytes, to replace endogenous iron losses, together with the recycling of some 20–25 mg of iron from effete erythrocytes via the reticulo-endothelial macrophages. In addition, there is normally a physiological reserve of iron in the hepatocytes (approximately 1 g) which further protects against imbalances in body iron homeostasis. The coordination and regulation of all of these processes has been ascribed to hepcidin, a 25-amino acid peptide hormone which is produced in the liver.1 2 Hepcidin expression is dramatically regulated by the levels of iron in the hepatic stores; expression is increased when liver iron is high and downregulated when the stores are depleted.3 In addition to modulation by liver iron content, hepcidin expression also responds dramatically to changes in the erythroid requirement for iron. Phlebotomy,4 haemolysis4 5 and elevated erythropoietin levels,4 major stimuli for reticulocytosis, all inhibit hepcidin production, and result in increased iron assimilation from the diet. Furthermore, hepcidin expression is increased in a number of chronic diseases and by inflammation2 4 6 indicating that pathological imbalances in hepcidin levels have severe consequences for body iron metabolism.
The cellular targets and the mode of action of hepcidin have been the subject of numerous recent studies. In vitro work with transfected cell lines suggests that hepcidin interacts directly with the ferroportin transporter resulting in its internalisation and degradation.7–9 These effects are rapid with maximal inhibition achieved 1–4 h following exposure to hepcidin.7 However, to date, it is not apparent whether this represents a global mechanism for the action of hepcidin in all ferroportin-expressing cells. In order to address this important issue, we have employed two well-characterised human cell lines commonly used in iron metabolism studies, namely the THP-1 monocyte/macrophage line and the intestinal epithelial Caco-2 cell. Cells were either co-cultured with the hepcidin-producing HuH7 hepatoma cell line10 or were exposed to a synthetic hepcidin peptide prior to measurements of ferroportin expression by using western blotting.
Functional data reveal that, once released, hepcidin acts as a negative regulator of both intestinal iron absorption11 12 and macrophage iron release10 13 suggesting that these tissues are the primary targets for hepcidin. Recent work has demonstrated that the in vivo effects of hepcidin are also rapid and can lead to hypoferraemia in mice within 1 h of a hepcidin injection.14 Interestingly, the maximal reduction in serum iron is evident some 2–4 h post-injection suggesting that the time course for the actions of hepcidin is similar both in vitro and in vivo. To determine whether the response to hepcidin was comparable in vitro and in vivo, we injected hepcidin into male C57BL/6 mice and measured changes in ferroportin protein expression in the spleen (an abundant source of reticulo-endothelial macrophages) and the duodenum. In addition we determined the effects of hepcidin treatment on functional outcomes by measuring changes in serum iron levels and duodenal iron transport.
METHODS AND MATERIALS
Cell culture
To achieve a fully differentiated monolayer, Caco-2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, for 21 days on Transwell inserts (Corning–Costar, High Wycombe, UK).15 HuTu80 cells (human duodenal epithelial cells) and HT29 cells (human colonic epithelial cells) were grown in the same medium and used for experiments upon reaching confluence. THP-1 cells, grown in RPMI 1640 medium containing 10% fetal bovine serum, were seeded at 5×105 cells per well on Transwell filters and treated with phorbol myristate acetate (100 nmol/l) to induce differentiation and filter attachment.
The day prior to experimentation, cells were placed in serum-free medium. The next morning the culture medium was replaced with fresh serum-free medium containing a commercially available synthetic hepcidin peptide (1 μmol/l; Peptides International Inc. Louisville, KY) for up to 24 h. This hepcidin peptide has been used successfully in a number of other studies.9 11 16
Co-culture
HuH7 hepatoma cells were grown in six-well plates in DMEM containing 10% fetal bovine serum and were used for experiments at 80% confluence. On the day of the experiments, cells were stimulated with interleukin-6 (IL6, 10 ng/ml; PeproTech EC Ltd, London, UK). HuH7 cells were overlaid with Transwell inserts containing either THP-1 cells or Caco-2 cells 2 h post-addition of IL6 and co-cultured for a further 4 or 24 h.
Animals and treatments
All procedures were approved by the Local Animal Ethics committee and were carried out in accordance with current UK Home Office legislation. Male C57BL/6 mice aged 4 weeks were housed in the Comparative Biology Unit at UCL (Hampstead campus) and were fed a diet containing 44 mg Fe/kg diet for 3 weeks prior to experimentation. During this time, animals were allowed ad libitum access to water. On the day of study, mice were given a single intraperitoneal injection of either 10 μg hepcidin or an equivalent volume of the diluent (saline) and used for experiments 4 h later.
Blood was collected by cardiac puncture from all animals at the end of the experimental period to measure serum iron and transferrin saturation using a commercially available assay (Randox Laboratories Ltd, Antrim, UK).
In vivo iron uptake
Animals were anaesthetised with pentobarbitone sodium (60 mg/kg, i.p.) and the duodenum was washed through with warm 0.15 mol/l NaCl followed by air. The lower end of the duodenal segment (proximal to the ligament of Treitz) was tied off and uptake buffer (0.15 mol/l NaCl, 10 mmol/l Hepes, pH 6.5) containing 0.2 mmol/l 59Fe2+ complexed with 4 mmol/l ascorbate was instilled proximally from a tied-in syringe. After 10 min incubation, during which time each animal’s body temperature was maintained at 37°C with a thermostatically controlled heating blanket, blood samples and the duodenal segment were removed. The duodenal lumen was emptied of its contents and was flushed thoroughly with uptake buffer containing 2 mmol/l unlabelled iron, followed by 0.15 mol/l NaCl, and finally dried overnight. The mucosa was weighed and, together with weighed blood samples and the animal carcass, counted in a gamma counter for determination of 59Fe activity. Mucosal iron retention, mucosal iron transfer and total mucosal iron uptake were calculated as described previously.11
Western blotting
Total plasma membranes from tissue samples and from cultured cells were prepared as described previously.17 18 All procedures were performed at 4°C. Briefly, the duodenal mucosa was separated from its underlying musculature by scraping with a glass slide. Splenic tissue was rinsed with PBS and chopped into fragments prior to homogenisation. Cultured cells were removed from Transwell plates using cell scrapers. Tissue and cell samples were homogenised in phosphate buffered saline (PBS) containing protease inhibitor cocktail (Sigma–Aldrich, Dorset, UK) with an Ultra Turrax homogeniser (2×30 s pulses on full speed). The ensuing homogenates were subjected to a brief centrifugation step (1500×g for 5 min) to precipitate unbroken cells and nuclei and the resulting supernatant was centrifuged for 30 min at 18 000×g to give a total plasma membrane fraction which was resuspended in PBS containing protease inhibitor cocktail (10 μg/ml), 1% NP-40 and 0.1% Triton X-100.
Membranes (40 μg) were solubilised in sample loading buffer and subjected to SDS-PAGE. Following immobilisation on nitrocellulose, the proteins were exposed to a commercially available anti-ferroportin antibody (1:1000 dilution, Alpha Diagnostics Inc., San Antonio, TX). Cross-reactivity was observed using an HRP-linked secondary antibody (Dako, Cambridgeshire, UK) and ECL Plus (GE Healthcare, Buckinghamshire UK). Band densities were semi-quantified using Scion Image software (Scion Corporation, Frederick, MD). At the end of the experiment, the nitrocellulose membranes were stripped (Western Stripping Buffer, Perbio Science Ltd, Northumberland, UK) and re-probed with an anti-actin antibody (1:2000 dilution, Sigma–Aldrich) which acted as a loading control.
Hepcidin peptide production by HuH7 cells in response to IL6 was assessed using an anti-hepcidin antibody (1:500 dilution) produced in-house against a synthetic peptide corresponding to the 25 amino acid residues of the mature form of human hepcidin. This antibody has been used previously to detect hepcidin protein in human tissue samples.19
Immunofluorescence
Tissues from mice injected with hepcidin or saline were perfusion-fixed in situ prior to harvesting. Briefly, the abdominal aorta of each anaesthetised animal was cannulated and each mouse was perfused with Hanks balanced salt solution (HBS, Sigma–Aldrich) to remove blood, followed by perfusion with HBS containing 2% paraformaldehyde. The spleen and duodenum from each animal were removed, embedded in OCT compound (Merck, Dorset, UK) and frozen in isopentane, pre-cooled in liquid nitrogen and stored at −80°C until use.
Cryostat sections (7 μm) of spleen and duodenum were mounted onto slides coated with poly-l-lysine (Sigma–Aldrich) and air-dried for 1 h. Sections were washed for 5 min in 0.1 mol/l PBS (pH 7.4) and then blocked for 30 min with PBS containing 10% normal goat serum (Sigma–Aldrich) and 1% Triton X-100. Sections were briefly washed with PBS and incubated overnight at 4°C with ferroportin antibody (1:100 dilution). Following overnight incubation, sections were washed (3×10 min, 0.1 mol/l PBS) and then incubated with FITC-conjugated swine anti-rabbit IgG (Dako) (1:100 dilution) for 1 h at room temperature. Subsequently, sections were washed (3×10 min, 0.1 mol/l PBS) and mounted with Vectashield mounting medium containing propidium iodide (Vector Laboratories Ltd, Peterborough, UK). Images were subsequently captured using a Leica TCS laser scanning confocal microscope and the manufacturer’s dedicated software.
Quantitative PCR
Total RNA was isolated from HuH7 cells using Trizol reagent (Invitrogen, Paisley, UK) according to the manufacturer’s instructions. Following first-strand cDNA synthesis, expression levels of hepcidin mRNA and 18S mRNA (used as a housekeeper), were analysed by real-time PCR using an ABI Prism 7000HT PCR cycler, using previously published primer sequences.12 20 and a Quanti-Tect SYBR Green PCR kit (Qiagen Ltd, West Sussex, UK) according to the manufacturer’s protocol. A quantitative measurement of each gene was derived from a standard curve constructed from known concentrations of PCR product and data are expressed as the hepcidin/18S ratio.
Statistics
Data are presented as mean ± SEM. Statistical differences (p<0.05) between groups were determined using either the Student unpaired t test or one-way analysis of variance followed by Tukey’s post-hoc test, where appropriate, using SPSS statistical package (SPSS UK Ltd, Surrey, UK).
RESULTS
Effect of hepcidin on ferroportin levels in THP-1 macrophages and intestinal epithelial Caco-2 cells
Previous work has shown that IL6 stimulates HuH7 cells to generate hepcidin, which can subsequently inhibit iron release from THP-1 cells.10 We exploited this approach to investigate the effects of hepcidin on the expression of ferroportin in the two major cell types controlling body iron metabolism, namely the macrophage and the intestinal epithelial cell. For these studies, we utilised two human cell lines, the THP-1 monocyte/macrophage line and the enterocyte-like Caco-2 cell, both of which are well characterised models that have been used extensively in studies on iron metabolism.
Addition of IL6 resulted in a rapid increase in hepcidin mRNA that was maximal after 2 h and remained elevated, compared with unstimulated cells, for the duration of the study (fig 1A). Increased levels of hepcidin protein were also detected in HuH7 cell lysates both 4 h and 24 h post-exposure to IL6 (fig 1B). For subsequent studies on ferroportin expression we pre-incubated HuH7 cells with IL6 for 2 h prior to co-culture with Transwell inserts containing either THP-1 cells or Caco-2 cells. In THP-1 cells, ferroportin protein levels were decreased significantly after 4 h of co-culture with HuH7 (p<0.01). Ferroportin expression remained depressed after 24 h exposure to IL6 (−21%); however, this was not statistically different from the levels observed in the untreated controls (fig 1C). In contrast to the THP-1 data, co-culture of Caco-2 cells with IL6-stimulated HuH7 cells did not significantly alter ferroportin protein levels at either the 4 h or 24 h time points (fig 1D).

To confirm that the observations with the co-culture model were due to hepcidin generated and released by HuH7 cells and not IL6, separate cultures of THP-1 and Caco-2 cells were incubated with IL6 alone for 4 or 24 h. In the absence of the hepcidin-generating HuH7 cell line there was no significant effect of IL6 on ferroportin expression in either THP-1 cells (fig 2A) or Caco-2 cells (fig 2B).

In subsequent studies as a positive control we incubated both THP-1 cells and Caco-2 cells with a commercially available synthetic hepcidin peptide. This manoeuvre reproduced the effects on ferroportin expression observed in the co-culture model, namely a rapid and significant downregulation of ferroportin protein levels in THP-1 cells occurring within 1–4 h of exposure to the peptide (fig 3A). Interestingly, in the studies with synthetic hepcidin, THP-1 cell ferroportin expression was still significantly decreased at 24 h. This apparent variance from the data from the co-culture study may have occurred due to differences in either the concentration, the half-life or the biological activity of endogenous versus synthetic hepcidin.
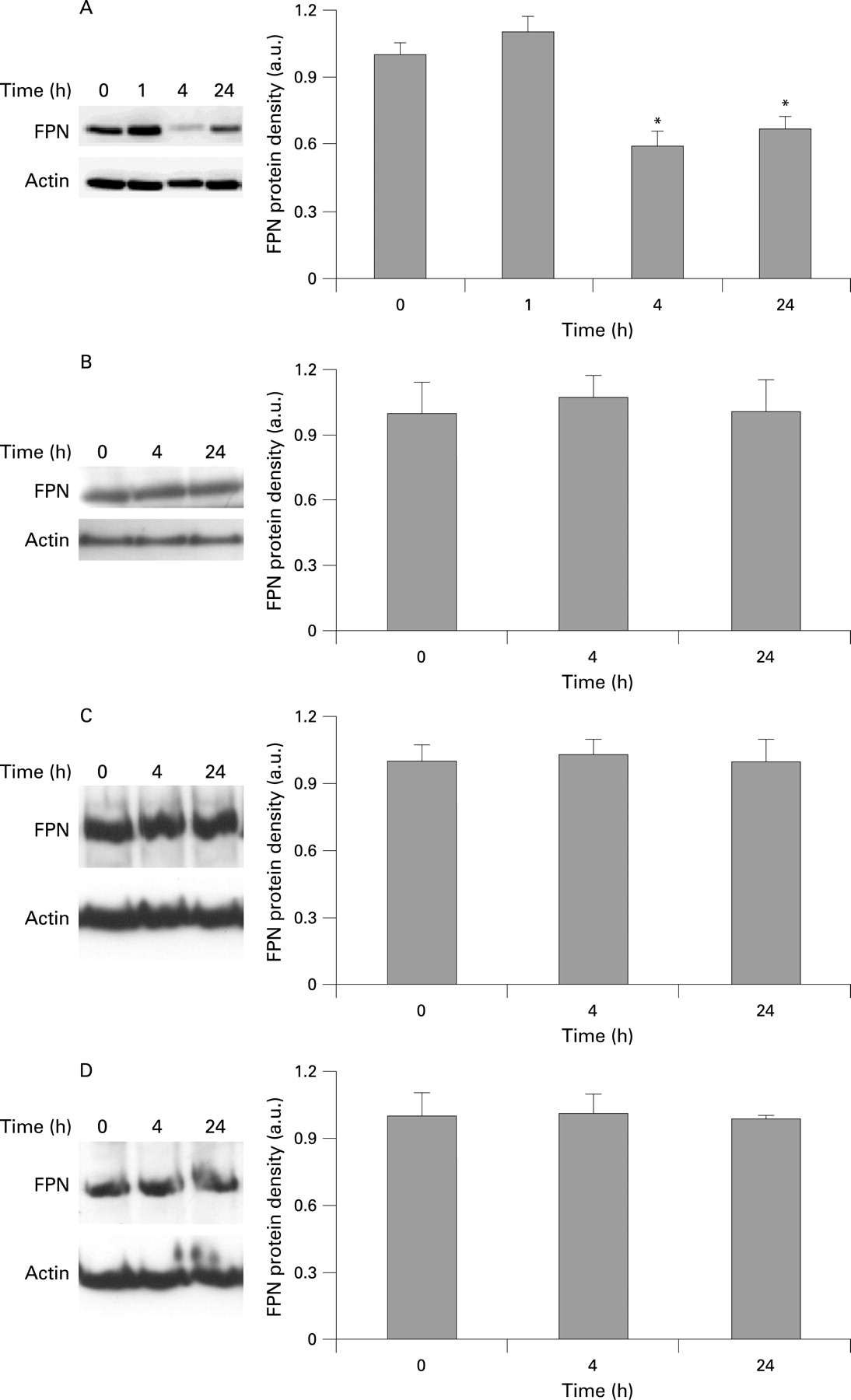
In keeping with the co-culture studies, there was no significant change in Caco-2 ferroportin expression following exposure to the synthetic hepcidin peptide (fig 3B). To ensure that this was not just a peculiarity of Caco-2 cells, two further human intestinal epithelial cell lines were also treated with hepcidin. In agreement with the Caco-2 cell data, ferroportin protein expression in duodenal HuTu80 cells (fig 3C) and HT29 colonic epithelial cells (fig 3D) was unaltered following exposure to hepcidin for 4 or 24 h.
Effect of hepcidin on ferroportin levels in spleen and duodenum
Taken together the in vitro data above suggests that hepcidin, either produced endogenously by HuH7 cells or provided as a synthetic peptide, causes a rapid macrophage-specific reduction in ferroportin protein expression. To determine whether these rapid effects of hepcidin were also evident in vivo we injected male C57BL/6 mice with the synthetic hepcidin peptide used the cell culture studies. After 4 h (the time taken to see the maximal effect of hepcidin on ferroportin expression in THP-1 cells) we isolated the spleen (an abundant source of iron recycling reticulo-endothelial macrophages) and the duodenum from control and hepcidin-treated mice. Ferroportin protein expression was assessed in both tissues by immunofluorescence and measured by semi-quantitative western blotting.
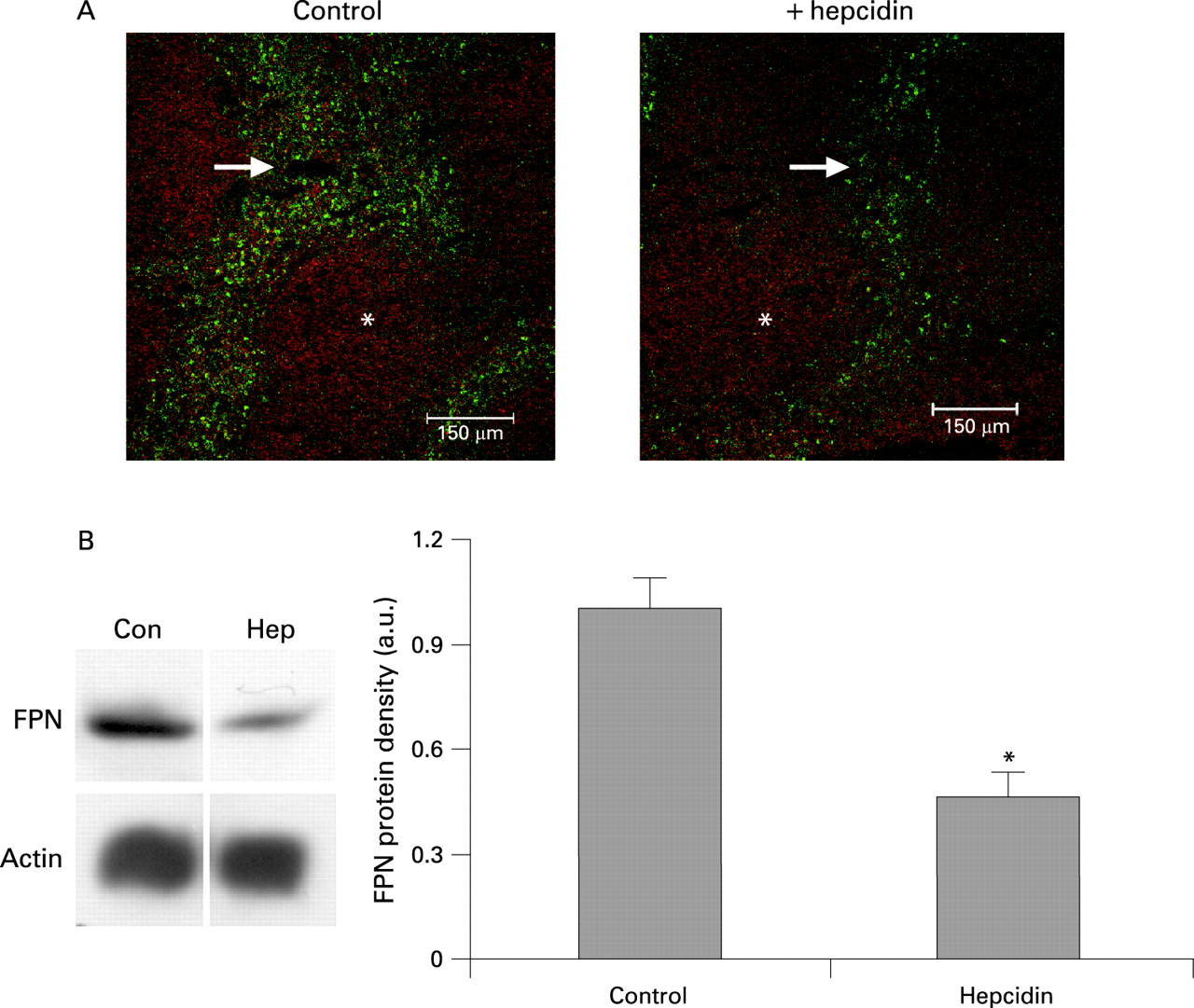
In the spleen, ferroportin immunoreactivity was associated with the macrophage-rich red pulp and was substantially reduced in the hepcidin-treated animals (fig 4A). Quantitative analysis of ferroportin levels in splenic tissue using western blotting was in agreement with the immunofluorescence data and revealed a 60% (p<0.001) reduction in ferroportin expression in hepcidin-treated mice compared with the saline-treated controls (fig 4B).
In the duodenum, ferroportin was localised to the epithelial layer. In addition, a number of strongly stained ferroportin-positive cells (most probably macrophages) were also noted in the lamina propria. Ferroportin immunoreactivity in the duodenal enterocyte layer was not altered following hepcidin treatment (fig 5A). However, the ferroportin-positive cells that were evident in the lamina propria of the control animals were absent from the hepcidin-treated mice. This may account in part for the small but statistically insignificant decrease (p>0.28) in ferroportin expression observed in the hepcidin-treated animals (fig 5B).

Serum iron levels in hepcidin-treated mice
A major consequence of hepcidin release is the inhibition of iron recycling from reticulo-endothelial macrophages (reviewed by Ganz21). Therefore, to determine whether there was an association between the decreased ferroportin expression in the macrophage-rich red pulp of the spleen and serum iron levels we collected blood from control and hepcidin-treated mice. In agreement with the proposed actions of hepcidin, we observed a substantial decrease in serum iron content in mice injected with the hepcidin peptide (control, 14.7 (SE 0.9) μmol/l; hepcidin-treated, 5.7 (SE 2.4) μmol/l; p<0.01), which was accompanied by a 40% decrease in transferrin saturation in the hepcidin-treated mice [control, 60.7% (SE 9.3)%; hepcidin-treated, 34.0% (SE 10.2%); p<0.1].
Short-term exposure to hepcidin does not alter duodenal iron transport
As well as controlling the rate of iron release from reticulo-endothelial macrophages into the circulation, hepcidin also acts on duodenal enterocytes to regulate the rate of dietary iron absorption.11 Therefore, an in vivo uptake technique was employed to determine the rapid effects of hepcidin on duodenal iron transport. Despite the dramatic hepcidin-mediated decrease in serum iron levels, duodenal iron transport was not altered in hepcidin-treated mice (fig 6). The absence of a rapid effect of hepcidin on duodenal iron transport was fully consistent with both the in vivo and in vitro observations that ferroportin expression was not compromised by incubation with hepcidin at this time point.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
Hepcidin is recognised as a major regulator of body iron metabolism. A number of in vitro studies have demonstrated that hepcidin regulates cellular iron homeostasis by controlling ferroportin expression.7–9 13 16 In this model, hepcidin is believed to bind directly to ferroportin and rapidly (within 1–4 h) induce its internalisation and degradation.7 This, in turn, impairs the release of iron from its target cells (namely the reticulo-endothelial macrophages and the duodenal enterocytes) into the circulation. In support of this hypothesis, a recent study in which radiolabelled hepcidin was injected into mice revealed that the peptide concentrates in tissues expressing the highest levels of ferroportin, i.e. the spleen, liver and duodenum.14
With the exception of one study16 all the data supporting the hepcidin–ferroportin model have been generated in cells where exogenous ferroportin has been over-expressed. Therefore, in the present investigation, using recognised human cellular models for macrophages (THP-1 cells) and intestinal enterocytes (Caco-2 cells) we have studied the effects of hepcidin on endogenous ferroportin expression.
THP-1 ferroportin levels were decreased significantly within 4 h of exposure to hepcidin (presented either as conditioned medium from IL6-stimulated HuH7 cells or as a synthetic peptide). Similar findings following hepcidin treatment have also been observed in the mouse J774 macrophage cell line13 and in mouse bone marrow derived macrophages.16 To confirm that these rapid effects of hepcidin were also evident in vivo we injected mice with hepcidin and 4 h later we isolated the spleen (a rich source of reticulo-endothelial macrophages) from hepcidin-treated mice and measured ferroportin levels. In agreement with the THP-1 data, ferroportin expression in the spleen was largely associated with the macrophage-rich red pulp, and transporter protein levels were significantly decreased in the hepcidin-treated mice. Furthermore, this decrease in splenic ferroportin expression was associated with a dramatic decrease in serum iron levels. This latter finding is consistent with a number of studies that have reported an association between elevated hepcidin levels in vivo and subsequent hypoferraemia.5 6 14 22 23 Taken together all of these studies support the published model for the cellular actions of hepcidin7 in which hepcidin binds to ferroportin in macrophages, decreases cell surface expression of the transporter and thereby inhibits iron release into the circulation resulting in hypoferraemia.
Along with the spleen, the duodenum is one of the major sites of ferroportin expression24–26 and as such would be expected to respond in a similar fashion to a hepcidin challenge. However, in contrast to the rapid effects of hepcidin in THP-1 macrophages and mouse spleen, ferroportin levels in three human intestinal epithelial cells line (Caco-2; HuTu80 and HT29) and in mouse duodenum were not altered over the same time scale. Similarly, there was no effect of acute hepcidin administration on duodenal iron transport. This contrasts with previous studies by our group11 12 and by others,27 which have reported decreased intestinal iron transport following chronic exposure to hepcidin.
The basis for this apparent differential sensitivity of the macrophage and intestinal epithelial cell to hepcidin is not fully clear. Previous work has shown that the effects of hepcidin on serum iron levels are dose-dependent14 and therefore one possibility is that the duodenum may simply require a higher concentration of hepcidin to elicit a response. However, our previous data would suggest that this is not the case since there is no impairment in duodenal iron transport in animals injected with 50 μg hepcidin 4 h previously (see Table 2 in Laftah et al11). Furthermore, data from our in vitro work with Caco-2 cells presented here, together with our previous findings,12 show no evidence for a dose-dependent effect of hepcidin on ferroportin expression.
A second possibility is that there are tissue-specific differences in the temporal kinetics of the response to hepcidin, i.e. the macrophage responds faster than the enterocyte. Our previous data support this option. In mice given multiple injections of hepcidin over a 24–72 h period there is a decrease in duodenal iron transport.11 In addition, Caco-2 cells incubated with hepcidin over a similar time course also display decreased iron transport.12 27 This mechanism is further supported by a recent study with transgenic mice containing an inducible hepcidin vector, which exhibit a significant decrease in duodenal ferroportin expression following chronic hepcidin exposure for 3 weeks.28
In addition to kinetic factors, we also believe that the basal level of hepcidin is important in determining whether the duodenum is responsive to hepcidin. There is clear evidence that in situations where there is a marked hepcidin deficiency, e.g. in iron deficiency,29 haemolytic anaemia,5 hypoxia30 USF2-null mice,31 and chronic alcohol loading,32 intestinal iron transport and/or ferroportin transporter expression are significantly elevated. Furthermore, in haemochromatosis there is a close correlation between hepcidin levels, which are inappropriately low considering the degree of iron loading, and duodenal iron absorption, which, in turn, is inappropriately high (reviewed by Fleming and Britton33).
The existence of different ferroportin protein species34 and splice variants35 has been postulated in various cell types and it is possible that there may be subtle differences in the way these putative ferroportin isoforms respond to a hepcidin challenge. Obviously, further studies would need to be performed to establish the credibility of such a mechanism.
Finally, in addition to macrophages and intestinal epithelial cells, ferroportin is also crucial for cellular iron homeostasis in the liver.36 Interestingly, some recent studies have suggested that the regulation of hepatic ferroportin expression may be largely independent of changes in hepcidin levels.37 38 In agreement with these earlier reports, we could not detect any differences in ferroportin expression in either liver membranes prepared from mice injected with hepcidin or in HuH7 human hepatoma cells exposed to the peptide compared with the respective untreated control groups (data not shown). Taken together with the intestinal data presented here, our work suggests that the iron-recycling macrophages are the primary target for the actions of hepcidin.
In conclusion, our study has shown for the first time that a cell-type specific response exists following an acute increase in hepcidin levels. We propose that upon its release into the circulation the initial rapid effects of hepcidin are evident at the level of the recycling macrophage, resulting in downregulation of ferroportin protein levels. These changes in transporter expression result in hypoferraemia. The inhibitory effects of hepcidin on duodenal iron transport and enterocyte ferroportin levels are only evident following chronic exposure to hepcidin for some 24–72 h. Since the reticulo-endothelial macrophages recycle some 20–25 mg Fe/day from senescent red blood cells, compared with only 1–2 mg Fe/day assimilated from the diet by the duodenal enterocytes, we believe the fact that macrophages respond more acutely to a hepcidin challenge is fully consistent with their paramount importance in maintaining body iron homeostasis.
Acknowledgments
We would like to thank Ms Linda Churchill and Dr Scott Wildman, University College London, for their help with immunofluorescence studies. We thank Professor Andrew McKie and Dr Yemesi Latunde-Dada, King’s College London, for providing us with the HuTu80 and HT29 cells used in this study.
REFERENCES
Footnotes
Competing interests: None
Funding: This work was funded by grants from the Biotechnology and Biological Sciences Research Council and the University of London Central Research Fund. Bomee Chung is part funded by the Overseas Research Student (ORS) Award Scheme.
Linked Articles
- Digest