Article Text

Abstract
Background:E-cadherin is an adhesion molecule involved in tumour invasion/metastasis. Silencing of E-cadherin by promoter CpG methylation has been shown in both familial and sporadic gastric cancers. Helicobacter pylori is a class I carcinogen in gastric cancer.
Aims: This study was undertaken to investigate the association between methylation of E-cadherin and H pylori in gastric mucosa from dyspeptic patients, and in intestinal metaplasia and primary and metastatic adenocarcinoma from surgical specimens of patients with gastric cancer.
Methods:E-cadherin methylation was studied using methylation specific polymerase chain reaction in microdissected tissue from biopsies or surgical resection specimens. E-cadherin expression was studied by immunohistochemistry.
Results:E-cadherin methylation was present in 31% (11/35) of gastric mucosae from dyspeptic patients, and was associated with H pylori infection (p=0.002), but was independent of the age of the patient or presence or absence of gastritis. E-cadherin methylation was present in 0% (0/8) of normal mucosa, 57% (12/21) of intestinal metaplasias, and 58% (15/26) of primary and 65% (21/32) of metastatic cancers. E-cadherin methylation status was concordant in 92% (11/12) of intestinal metaplasias and primary cancers, and in 85% (17/20) of primary and metastatic cancers from the same resected specimen. E-cadherin methylation in gastric cancer was associated with depth of tumour invasion (p=0.02) and regional nodal metastasis (p=0.05).
Conclusion:E-cadherin methylation is an early event in gastric carcinogenesis, and is initiated by H pylori infection.
- E-cadherin methylation
- gastric cancer
- intestinal metaplasia
- Helicobacter pylori
- PCR, polymerase chain reaction
- MSP, methylation specific polymerase chain reaction
- COX-2, cyclooxygenase 2
Statistics from Altmetric.com
- PCR, polymerase chain reaction
- MSP, methylation specific polymerase chain reaction
- COX-2, cyclooxygenase 2
E-cadherin is a transmembrane glycoprotein expressed on epithelial cells, and is responsible for calcium dependent homotypic cell adhesion. It binds to several cytosolic proteins including α-, β-, and γ-catenins, which are in turn linked to actins.1,2 Cell-cell and cell-matrix interactions are crucially involved in neoplastic transformation and metastasis.3–6 Defective cell adhesion may contribute to loss of contact inhibition of growth, which is an important early step in the neoplastic process. Furthermore, loss of cell adhesion may account for the ability of cancer cells to cross normal tissue boundaries and metastasise. The importance of E-cadherin in maintaining cell adhesion implies that its dysfunction may play an important part in tumorigenesis.7
In gastric carcinogenesis, the critical role of E-cadherin is underlined by the observation that familial gastric cancer is related to germline mutation of the E-cadherin gene.8,9 Furthermore, somatic mutations of E-cadherin were found in approximately 50% of gastric carcinoma of the diffuse histological type.10 Finally, in two kindreds with familial gastric cancer and germline E-cadherin mutation, promoter CpG hypermethylation was found to be the second “genetic hit” in abrogating E-cadherin expression.11 These results show that E- cadherin is an important putative tumour suppressor gene involved in gastric carcinogenesis.
Gastric carcinogenesis is a multistep process with morphological progression involving multiple genetic and epigenetic events, and an intestinal metaplasia-dysplasia-invasive carcinoma sequence exists.12 It is now increasingly recognised that epigenetic silencing of gene expression by promoter CpG hypermethylation is an important alternative mechanism in inactivating tumour suppressor genes and tumour associated genes in cancers,13 and in gastric cancers and precursors.14,15Helicobacter pylori infection is an important aetiological risk factor in gastric cancer, and has been classified as a group I or definite carcinogen by the World Health Organization’s International Agency for Research on Cancer.16
In this study, we evaluated the role of E-cadherin methylation in dyspeptic patients without metaplasia or dysplasia, and in sporadic gastric carcinomas, by studying promoter hypermethylation of the gene in gastric mucosa in patients without cancer, in intestinal metaplasia, and in primary carcinoma and metastatic lymph nodes from patients undergoing surgery for gastric cancer. In particular, the association between E-cadherin methylation and H pylori was assessed.
MATERIALS AND METHODS
Patients and specimens
Thirty five gastric mucosal biopsies obtained from patients undergoing upper endoscopy for dyspepsia were studied. Haematoxylin and eosin stained slides were evaluated for the presence of gastritis, intestinal metaplasia, or dysplasia. H pylori infection was evaluated by histology and urease breath test. DNA from eight normal mucosae, 21 intestinal metaplasias, and 26 primary adenocarcinomas and 32 metastatic lymph nodes from gastrectomy specimens of patients undergoing resection for gastric adenocarcinoma were studied. The tumours were staged according to the Japanese Research Society for Gastric Cancer17 and classified according to the World Health Organization classification scheme.18 DNA was obtained by microdissection from 5 μm thick haematoxylin and eosin stained paraffin embedded tissue sections without coverslip, as described previously.19H pylori infection status was evaluated in 18 primary adenocarcinoma specimens by histology. Informed consent for tissue procurement was obtained from all patients.
Methylation specific polymerase chain reaction (MSP) for E-cadherin promoter methylation
The methylation status of the E-cadherin promoter was determined by bisulphite treatment of DNA followed by MSP, as described previously.20 Briefly, 2 μg of DNA were denatured with 2 M NaOH at 37°C for 10 minutes, followed by incubation with 3 M sodium bisulphite, pH 5.0, at 50°C for 16 hours. Bisulphite treated DNA was then purified (DNA Cleanup Kit; Promega, Madison, Wisconsin, USA), incubated with 3 M NaOH at room temperature for five minutes, precipitated with 10 M ammonium acetate and 100% ethanol, washed with 70% ethanol, and resuspended in 20 μl of distilled water. DNA (2 μl) was then amplified by polymerase chain reaction (PCR) with two sets of primers specific for the methylated and unmethylated alleles, as described by Herman and colleagues20 and Graff and colleagues21 (fig 1). CpGenome Universal Methylated DNA (Intergen, Purchase, New York, USA) in which E-cadherin was methylated and reagent blanks were used as positive and negative controls in each experiment. All tests were performed in duplicate. For confirmation of the specificity of MSP, PCR products from the methylated and unmethylated primers were gel purified and sequenced, as previously described.22

(A) Confirmation of the specificity of methylation specific polymerase chain reaction (MSP). The wild-type E-cadherin gene sequence is shown together with the sequence of the methylated polymerase chain reaction (PCR) product. M1: PCR product (shown here from nucleotides 2280 to 2339, Genbank accession No D49685) with first primer set, as described by Herman and colleagues,20 for the methylated E-cadherin allele (forward primer: 5′- TTA GGT TAG AGG GTT ATC GCG T -3′; reverse primer: 5′- TAA CTA AAA ATT CAC CTA CCG ACC -3′). Primers for the unmethylated E-cadherin allele were forward: 5′- TAA TTT TAG GTT AGA GGG TTA TTG T -3′; reverse: 5′- CAC AAC CAA TCA ACA ACA CA -3′. In the MSP product, methylated cytosine residues that are unchanged are underlined in blue, and the unmethylated cytosine residues that have become uracil/thymidine are underlined in red. (B) M2: PCR product from second primer set for methylated E-cadherin allele (shown here from nucleotides 2229 to 2283), as described by Graff and colleagues21, for the methylated E-cadherin allele (forward primer: 5′- GTG GGC GGG TCG TTA GTT TC -3′; reverse primer: 5′- CTC ACA AAT ACT TTA CAA TTC CGA CG -3′). Primers for the unmethylated E-cadherin allele were forward: 5′- GGT GGG TGG GTT GTT AGT TTT GT -3′; reverse: 5′- AAC TCA CAA ATC TTT ACA ATT CCA ACA -3′.
Immunohistochemical staining for E-cadherin
E-cadherin expression was examined by immunostaining using the avidin-biotin complex immunoperoxidase method.23 Briefly, 4 μm thick tissue slides were deparaffinised in xylene and rehydrated serially with alcohol and water. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 10 minutes, followed by microwave antigen retrieval for nine minutes at 95°C in 10 mM sodium citrate buffer, pH 6.0. The slides were then incubated with an avidin conjugated monoclonal anti-E-cadherin antibody (HECD-1 1:500 dilution in phosphate buffered saline; Zymed Laboratories Inc., South San Francisco, USA) in a moist chamber at 37°C for one hour. Bound antibody was detected by a biotinylated secondary antibody and the avidin-biotin complex immunoperoxidase method (Dako Corp., Carpinteria, USA). The slides were finally counterstained with Mayer’s haematoxylin. For a negative control, the primary antibody was replaced with mouse IgG. Slides with normal gastric mucosa were used as a positive control. Furthermore, positive E-cadherin staining in the adjacent non-involved gastric mucosa served as an internal positive control. E-cadherin staining was classified as absent staining, cytoplasmic distribution, heterogeneous staining, or normal membranous staining. Because staining patterns often varied within the same tumour, particularly when the degree of differentiation varied, the classification was based on the dominant pattern. Tumours with more than 10% variation were scored as heterogenous.
Statistical analysis
Fisher’s exact test and χ2 test were used to compare categorical associations and the Student’s t test for continuous associations. Two sided tests were used to calculate p values. Multivariate analysis using a forward conditional model was used to evaluate the contribution of age, gastritis, and H pylori infection to E-cadherin methylation in the gastric mucosae of patients with dyspepsia. The relationship between E-cadherin methylation status and clinicopathological features, including age, histological subtypes of the carcinoma, depth of tumour invasion, presence and extent of regional lymph node, and distant metastasis, were evaluated in the 26 primary cancers.
RESULTS
MSP for E-cadherin
MSP with both sets of primers for the methylated and unmethylated E-cadherin promoter gave expected results in the positive and negative controls, and concordant results in all of the tested specimens. Sequencing of the PCR products confirmed the expected nucleotide changes associated with bisulphite modification in the methylated and unmethylated alleles (fig 1A, 1B).
E-cadherin methylation, expression, and clinicopathological associations in gastric mucosae from patients with dyspepsia
The clinicopathological associations of 35 patients with dyspepsia are tabulated in table 1. Gastritis was present in 31 (89%) cases, and H pylori infection in 19 (54%) cases. Patients with H pylori infection were older compared with those without infection (53 (14) years v 38 (12) years; p=0.002). H pylori infection was also associated with gastritis (p=0.009). Intestinal metaplasia or dysplasia was not present in any biopsy on histological assessment. E-cadherin methylation in gastric mucosa was present in 11 (31%) patients (fig 2A, 2B). All 11 cases with E-cadherin methylation had gastritis (NS) (table 1). E- cadherin methylation was associated with H pylori infection: 53% (10/19) of patients with H pylori infection had E-cadherin methylation versus 94% (15/16) of patients without H pylori infection and no methylation (p=0.002) (table 1). Patients with E-cadherin methylation were older compared with those without methylation (53 (13) years v 43 (15) years; p=0.048). H pylori infection was the only factor associated with E-cadherin methylation by multivariate analysis (p=0.013 (95% confidence interval 0.007–0.55)). However, immunohistochemical staining showed normal membranous staining of E-cadherin in samples with or without E- cadherin methylation. This was probably due to the fact that immunohistochemical staining was not as sensitive as PCR in detecting subpopulations of cells with gene methylation and hence downregulation of E-cadherin.
Association of E-cadherin promoter methylation with age, gastritis, and Helicobacter pylori infection status in 35 patients with dyspepsia

(A) Analysis of methylation of E-cadherin in gastric mucosae from patients with dyspepsia. Samples N3–N6 are methylated, and samples N1, N2, and N7 are unmethylated. Methylation specific polymerase chain reaction (MSP) was performed with the first primer set, resulting in a methylated polymerase chain reaction (PCR) product of 116 bp and an unmethylated PCR product of 97 bp. (B) Analysis of methylation of E-cadherin in gastric mucosae from patients with dyspepsia, as shown in (A), but with MSP performed with the second primer set. Samples N3–N6 are methylated, and samples N1, N2, and N7 are unmethylated. (C) Analysis of methylation of E-cadherin in intestinal metaplasia (IM), gastric adenocarcinomas (T), and metastatic lymph nodes (LN) from patients with gastric cancer. Samples IM2, T1, T2, LN1, and LN2 are methylated, and sample IM1 is unmethylated. MSP was performed with the first primer set.
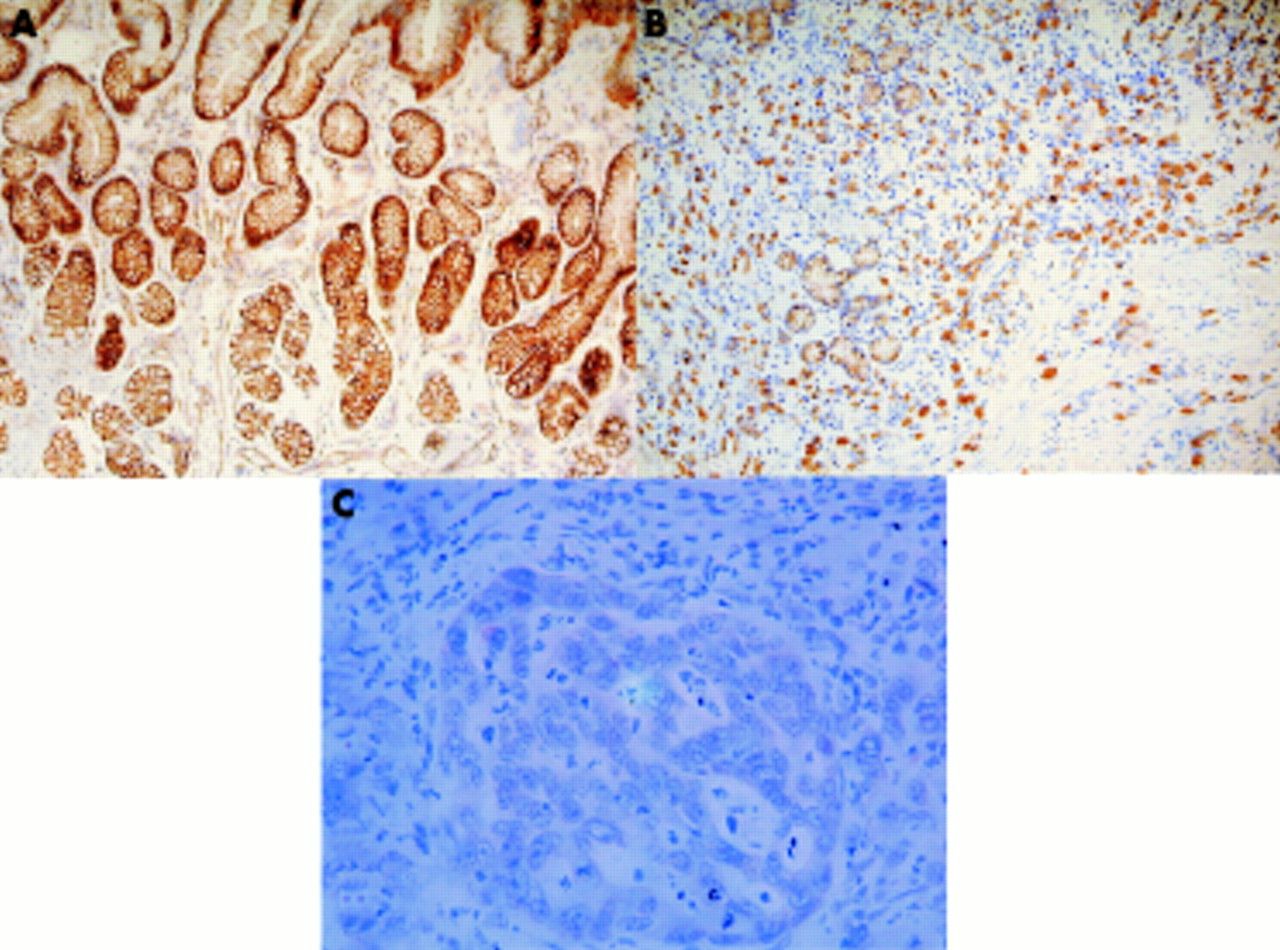
E-cadherin methylation, expression, and clinicopathological associations in resected specimens in patients with gastric cancer
E-cadherin methylation was present in 0 of 8 (0%) normal mucosae, 12 of 21 (57%) intestinal metaplasias, 15 of 26 (58%) primary cancers, and 21 of 32 (65%) metastatic cancers (fig 2C). Methylation status was concordant between 11 of 12 (92%) intestinal metaplasias and cancers, and 17 of 20 (85%) primary and metastatic cancers from the same patients. One patient had methylation in intestinal metaplasia but not primary cancer, another had unmethylated primary cancer but methylated metastatic cancer, and two patients had methylated primary cancer but unmethylated metastatic cancer. E-cadherin immunohistochemistry was performed on 13 primary and nine metastatic cancers. Eleven primary cancers had loss of membranous distribution of E-cadherin (fig 3A) and had E- cadherin methylation (100%). Of two primary cancers with retained E-cadherin expression, the gene was methylated in one case and unmethylated in the other. Seven metastatic cancers in the lymph node had diminished E-cadherin staining (fig 3B), and five (71%) had E- cadherin methylation. In the two remaining cancers with retained E-cadherin expression, the gene was methylated in one cancer and unmethylated in the other. E-cadherin methylation was associated with the depth of tumour invasion. All three tumours confined to the lamina propria (T1) were unmethylated but 15 of 23 (65%) tumours with invasion of the muscularis propria or beyond were methylated (p=0.02) (table 2). E-cadherin methylation was also associated with regional nodal metastasis. Four of five (80%) patients without nodal metastasis were unmethylated but 14 of 21 (67%) patients with regional metastasis were methylated (p=0.05) (table 2). However, methylation was not related to age, H pylori infection status, histological subtype, or distant metastasis status.
Clinicopathological associations of E-cadherin methylation in patients with gastric cancer

{kind=link}
{kind=link}
{kind=link}
Immunohistochemical staining for E-cadherin, showing (A) membranous distribution of staining in the normal gastric mucosa, (B) cytoplasmic staining in gastric cancer cells, and (C) cytoplasmic staining in neoplastic cells in a metastatic lymph node.
DISCUSSION
In this study, we found E-cadherin methylation in patients with H pylori infection without metaplasia or dysplasia, in intestinal metaplasia, and in primary and metastatic cancers from patients undergoing gastrectomy for gastric cancer. E-cadherin methylation was present in one third of gastric mucosae from patients with dyspepsia, and was associated with age and H pylori infection. However, by multivariate analysis H pylori infection was the only significant contributing factor. H pylori can induce transcriptional activation by inflammatory mediators such as nuclear factor κB and cyclooxygenase 2 (COX-2).24,25 Methylation in gastric epithelial cells may occur as a consequence of or as an adaptive protective response due to chronic exposure to inflammatory mediators overproduced during infection, a mechanism that has been described in viral infection,26 or in metaplasia, dysplasia, and carcinoma developing due to chronic reflux injury of the oesophagus and inflammatory bowel disease in the colorectal region.27,28 The former is supported by a previous study which showed that H pylori stimulated COX-2 expression in gastric cell lines without promoter methylation of the COX2 gene but not in cell lines with methylation of COX-2 promoter.25 However, COX-2 expression can be induced by demethylation treatment with 5-azacytidine followed by exposure to H pylori in cell lines with methylation of COX-2 promoter. It is therefore possible that E-cadherin methylation may be mediated through similar mechanisms in response to H pylori infection. Finally, it is intriguing to note that only about half of gastritis cases with H pylori infection showed E-cadherin methylation. This could be related to a sampling effect as E-cadherin methylation in relation to H pylori infection might be focal. Alternatively, other collaborative mechanisms together with H pylori infection may be involved in E-cadherin methylation.
Also, it is apparent that only a small proportion of H pylori related gastritis cases progress to carcinoma. According to Knudson’s “two hit” hypothesis,29 two genetic mutations are required for inactivation of a putative tumour suppressor gene. Promoter methylation with suppression of gene expression may collaborate with mutation or deletion to cause gene inactivation, thereby fulfilling Knudson’s hypothesis.30 Thus the occurrence of additional genetic events may be necessary for E-cadherin gene inactivation, leading to initiation of carcinogenesis.
We did not observe an association between H pylori infection and E-cadherin methylation in gastric cancers. This may be related to the fact that this was a retrospective analysis so that prior H pylori infection status was not known in all of the cases of cancer. However, it is also well known that H pylori infection declines with the development of gastric cancer31 and histological examination alone cannot reflect accurately previous exposure to H pylori infection.
E-cadherin methylation was present in 57% of intestinal metaplasias in patients with gastric cancers in our study. The timing of hypermethylation during tumour development may vary among different genes and tumour types. Kang and colleagues15 demonstrated that methylation at DAP-kinase, THBS1, and TIMP-3 was present in gastritis whereas p16 and hMLH1 were present in intestinal metaplasia and cancer. We demonstrated that the frequency of E-cadherin methylation was the highest compared with the methylation frequency at p16 (2.1%), hMLH1 (6.3%), THBS-1 (34.7%), DAP-kinase (36.7%), and TIMP-3 (36.7%) in intestinal metaplasia.15
Methylation of E-cadherin was present in 58% of primary cancers and 66% of cancers metastatic to lymph nodes in our study, a frequency comparable with previous reports.14 Interestingly, in the metastatic nodes without E-cadherin methylation, there were two cases where the primary tumour showed E-cadherin methylation. Similar findings have been observed in immunohistochemical studies of E-cadherin expression.23 It has been postulated that expression of E-cadherin might be dynamic. Thus downregulation of E-cadherin in the primary tumour may allow cell dispersal into the circulation while re-expression of E- cadherin may allow cell deposition at distant sites to form metastases.23
There was, in general, concordance between E-cadherin methylation and diminished E- cadherin expression by immunostaining. However, there was one primary and one metastatic cancer in a lymph node where E-cadherin methylation was not associated with diminished E- cadherin staining. This may indicate that cancer cells with E-cadherin methylation were only a minor subclone of the tumour, which was picked up by the sensitive MSP.
Clinicopathological correlations showed that E-cadherin methylation was associated with depth of invasion and nodal metastasis. This pattern suggests that increasing acquisition of E- cadherin methylation may be related to progressive tumour development. If E-cadherin methylation is also an important initiating event, as occurs in intestinal metaplasia progressing to gastric cancer, a quantitative increase in E-cadherin methylation may be expected as the neoplastic clone progressively increases in size. To further validate these possibilities, analysis of E-cadherin methylation by a quantitative assay is required.32 These biological considerations notwithstanding, depth of invasion and nodal metastasis in relation to E-cadherin methylation were parameters that may be associated with poor prognosis. In fact, the role of E-cadherin as a prognostic marker in gastric cancer has been previously shown by immunohistochemical studies23,33 and serum enzyme linked immunosorbant assays.34 Furthermore, loss of E-cadherin expression is most pronounced in epithelial carcinomas with an infiltrative growth pattern associated with no intercellular cohesion, such as invasive lobular breast cancer and diffuse-type gastric adenocarcinoma. The increased invasiveness of tumours with E-cadherin methylation identified in our study might be consistent with these findings.
In conclusion, we have shown an association between E-cadherin methylation in the stomach and H pylori infection in dyspeptic patients without cancer, and in intestinal metaplasia and cancer. Furthermore, E-cadherin methylation in gastric cancer was also associated with depth of tumour invasion and nodal metastasis in gastric adenocarcinoma. However, the results of this study do not exclude mechanisms other than H pylori infection that may lead independently to E-cadherin methylation. A working hypothesis may be that gastric mucosal infection by H pylori initiates E-cadherin methylation which may subsequently progress to intestinal metaplasia and invasive cancer. Such a hypothesis will require vigorous testing in future studies. Finally, the potential contribution of E-cadherin methylation to other H pylori related pathologies may warrant further investigation.
Acknowledgments
YLK is supported by the Kadoorie Charitable Foundation. The project was supported by the University of Medicine Department Research Grant.