Article Text

Microsatellites
Abstract
Microsatellite DNA sequences consist of relatively short repeats of one to five base pair units; together with satellites and minisatellites they comprise a larger family known as tandemly repetitive sequences. Microsatellites are found both in prokaryotes and eukaryotes, including humans, wherein they appear scattered almost at random throughout the genome. Although in prokaryotes distinct biological functions have been demonstrated, the role of microsatellites in eukaryotes is less clear. Nevertheless, several interesting hypotheses exist suggesting that certain microsatellites may exert subtle influences on the regulation of gene expression. Although the presence of these subtle mechanisms may be beneficial to a whole population, when they go wrong, as is thought to happen in the case of human trinucleotide repeat associated diseases, such as Huntington's disease, the consequences for the individual can be fatal. Most human microsatellites probably have no biological use at all; however, they are extremely useful in such fields as forensic DNA profiling and genetic linkage analysis, which can be used to search for genes involved in a wide range of disorders. With a primary focus on humans, it is the aim of this review to present an up to date discussion, both of the biological aspects and scientific uses of microsatellite sequences. In the latter case, basic theoretical and technical points will be considered, and as such it may be of use both to laboratory and non-laboratory based readers.
- microsatellites
- genome
- genetics
- DNA
Statistics from Altmetric.com
Microsatellites
Three billion may seem to be a very large number when used in any “normal” context. However, when used to refer to the number of repeating units of genetic information that are sufficient to encode the blueprint for something as complex as a human being, it never ceases to amaze me that this number is big enough! Consequently, it comes as even more of a surprise to find that most of these units appear to be “functionless junk”! In fact “functional DNA”, consisting of transcribed genes and regions involved either in transcriptional regulation or in maintaining chromosomal structure/integrity, is thought to comprise less than a sixth of the total human genome. This observation immediately raises a considerable number of questions, namely: what is this genetic junk made up of? Is it really junk—does it truly possess no useful function either directly or indirectly? How is it acquired? What other types of organisms possess it? And can we make any use of it?
Fully covering the questions raised above would provide more than ample scope for writing an entire book, and would extend well beyond the remit of this review. Nevertheless, even focusing as intended upon microsatellites and their relevance to humans, it should become clear that most of these questions remain ones that will require consideration, even if only in passing.
Although only about 3% of the human genome encodes expressed protein sequences, the inclusion of non-expressed regions with known function, as well as pseudogenes, recognisable gene fragments, and all intronic sequences brings the total to around 30%. Of the remaining 70%, about four fifths comprises sequences that appear to be unique or repeated only rarely (fig 1). Apart from the important use of providing what could be referred to as “evolutionary padding”, this DNA may truly be functionless. The residual fifth is moderately to highly repetitive, and can be divided into two types, depending on whether the individual repeat units are dispersed singularly (interspersed repetitive DNA) or clustered together (satellite DNA).
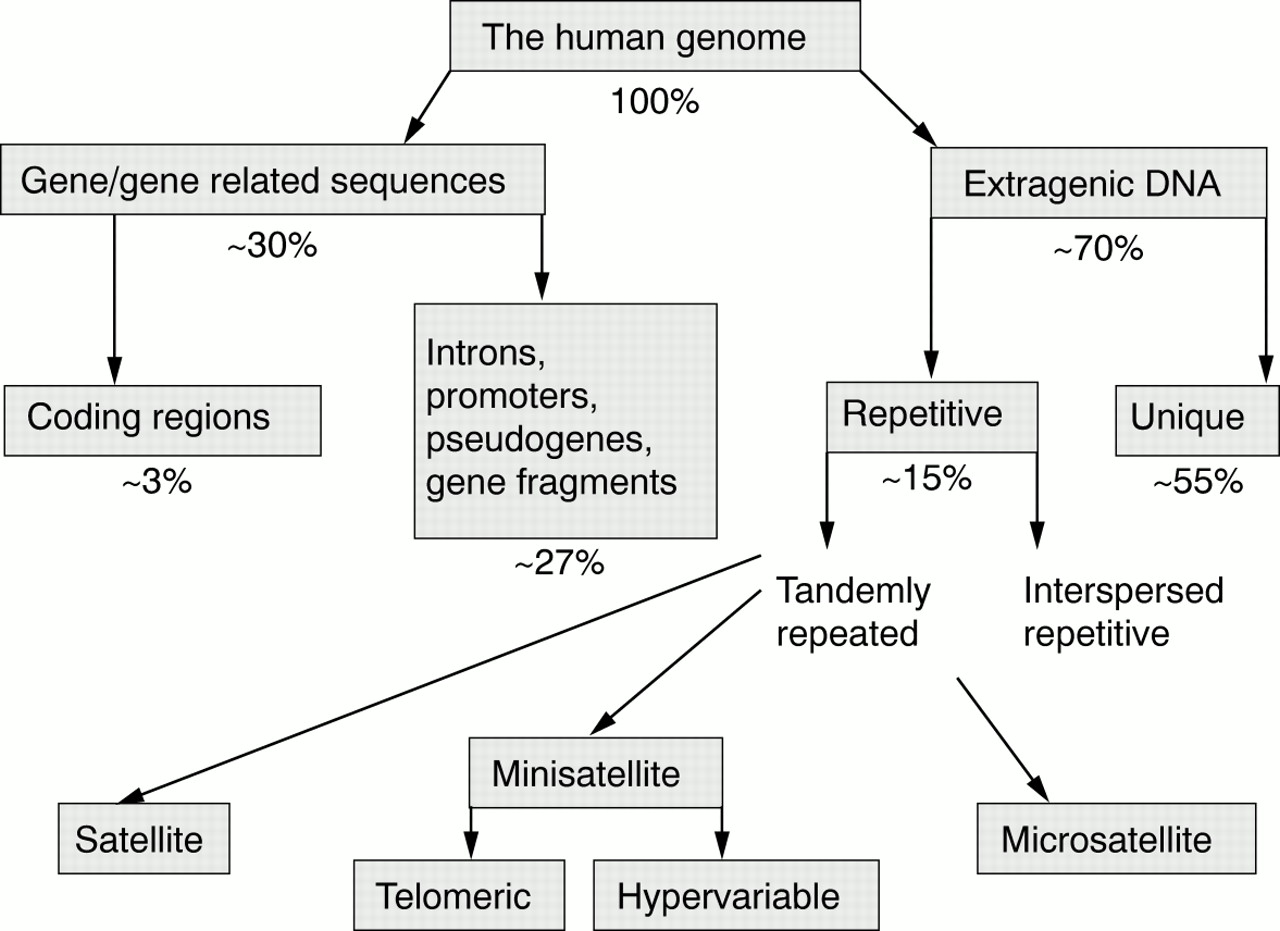
Sequential breakdown of the human genome into component DNA types.
The topic of interspersed repetitive DNA will not be discussed in detail within this review; however, a few interesting points are worthy of note before moving on to consider satellite DNA. There are several families of interspersed repetitive DNA, although the two largest are known as short and long interspersed nuclear elements (SINEs and LINEs, respectively),1 and it is to the SINE family that the frequently mentioned Alu repeat belongs. Alu is primate specific, and consists of a DNA sequence approximately 280 bp (base pairs) long, which is imperfectly repeated around three quarter of a million times within the human genome. This makes it the single most abundant human DNA sequence, occurring on average once every 4 kb (kilo base pairs), and accounting for more than 5% of total DNA. Alu sequences, in common with many interspersed repeats, appear to have arisen and subsequently been propagated by means of retrotransposition; that is to say they are genomic (human or viral) DNA sequences that have been transcribed into RNA before being reverse transcribed back into chunks of isolated DNA. Such chunks are then able to reinsert elsewhere within the human genome. Indeed some members of the LINE family of interspersed repeats are thought to encode functional reverse transcriptases and hence could be capable of facilitating their own propagation.2
Satellites
Like interspersed repetitive DNA, certain types of satellite sequences can be found widely dispersed throughout the genome. However, satellite DNA differs in that at any one locus the basic repeat unit is found as an array of tandem duplications. Indeed, the various types of satellite DNA are commonly referred to collectively as tandemly repeated DNA. Furthermore, for many satellites, the number of tandem repeats at a given locus can vary from one individual to the next. In short, satellite sequences are frequently polymorphic which, as we shall see later, can make them very useful for scientists. This apparent instability, in conjunction with their tandemly repeating nature, suggests that the origin of satellite DNA is likely to be very different to that of the interspersed repetitive DNA elements mentioned above.
Based upon both the size of the individual repeating unit and the overall repeat length, tandemly repeated DNA has been split into three subgroups, known as satellites, minisatellites, and microsatellites. Despite attempts,3 there are no universally accepted size limits to the boundaries of each subgroup, although the figures quoted by many authors are in general agreement, hence those used in this review represent the consensus (table 1).
Details of the different types of satellite DNA
Satellite DNA was the first of the tandemly repeating DNA sequences to be discovered,4 and was so named by its appearance as minor or “satellite” bands that separated from the “bulk” DNA upon buoyant density gradient centrifugation. Further satellite sequences were identified later by restriction digestion of genomic DNA; by using restriction enzymes that cut only once within the basic repeat unit, large amounts of DNA corresponding to the size of the monomer unit would be seen after electrophoretic separation. Indeed, as a result of incomplete digestion and random point mutations causing the loss of restriction sites within certain individual repeat units, a “laddering” effect owing to the presence of various multimers was often seen.5 The basic repeat unit of satellite DNA can vary from as little as five to as many as several hundred base pairs; however, the overall repeat size at any one locus is enormous, and may cover anywhere from around 100 kb to several Mb (mega base pairs). Human satellite DNA is not transcribed, and is found in heterochromatin (condensed areas of chromatin consistently lacking actively expressed genes), especially centromeric heterochromatin. However, there is no evidence to suggest that these restrictions upon chromosomal localisation are associated with any functional importance. Thus, the jury remains out on the extent to which satellite DNA constitutes true genetic junk. Furthermore, because of its enormous size and restricted localisation, satellite DNA is of no real use for either individual DNA profiling or genetic linkage studies.
Minisatellites
Unlike satellite DNA, minisatellites are more interesting and have been useful. Minisatellite DNA consists of moderate arrays of tandem repeats spanning approximately 100 bp to 20 kb in length. It can be subdivided into two types, the first of which is known as telomeric. Telomeric DNA consists of 10–15 kb of hexanucleotide repeats (mainly TTAGGG), added to the telomeres of all chromosomes by the enzyme telomerase. Such DNA is most definitely functional in that it protects the ends of chromosomes from degradation and provides a means for the complete replication of telomeric sequences. It is also thought to play a role in the pairing and orientation of chromosomes during cell division.
The second type of minisatellite sequences are hypervariable minisatellite DNA, and include those first discovered in 1985 by Alec Jeffreys and colleagues at Leicester University.6 The basic repeat unit may vary in length from six to > 50 nucleotides, with the overall number of repeats at any one locus usually being highly polymorphic between individuals. Also commonly known as variable number tandem repeats or VNTRs (despite the fact that by true definition, this term would also encompass satellite and microsatellite sequences), it is their highly polymorphic nature that makes minisatellites so useful in the field of DNA profiling. Initially, the technique developed by Jeffreys's team was known as DNA fingerprinting.7–10 This involved hybridising short synthetic probes containing common core sequences, such as GGGCAGGANG (which Jeffreys showed was found within most VNTRs), to an individual's genomic DNA, which had been digested with restriction enzymes and then separated by electrophoresis (a process known as Southern blotting). The probes would “stick” to dozens of DNA fragments, the sizes of which were determined by the number of repeats they contained. Consequently a complex banding pattern or “fingerprint” would be produced, which was essentially unique to that individual. Furthermore, offspring would display a pattern made up of equal combinations of the bands present in their parents' fingerprints. This technique started a revolution in fields such as forensic science and paternity testing, but still had some disadvantages. The principal problem was that there was no practical way of knowing which pairs of bands in the fingerprint represented alleles at a particular minisatellite locus; consequently, it was not possible to calculate allele frequencies and hence reliably state the odds of bands within a fingerprint matching as a result of chance. There were also potential technical difficulties and a considerable opportunity for human error. Thus, although doubtlessly remaining very useful, the DNA fingerprinting technique was less than ideal in a courtroom situation.
As knowledge of minisatellite sequences increased, and as a result of the problems described above, the so called multilocus minisatellite probes of DNA fingerprinting were soon replaced with specific single locus minisatellite or VNTR probes.11–14 This solved the main problem of fingerprinting and with, typically, the use of between four and 10 highly polymorphic VNTRs, astronomically large odds against matches being the result of chance were quoted in the courts. Looking back, these figures were probably over optimistic because they did not take sufficient account of factors like the variations in allele frequencies between different populations/ethnic groups or the raised odds of matches within genetically isolated communities. Nevertheless, the odds of genuine chance matches were almost certainly far outweighed by the odds of matches being incorrectly scored as the result of human error. Most of the potential for human error lay within a process known as “binning”. This was where a decision had to be made as to which allele (or repeat number) corresponded to which band on the Southern blot autoradiograph. Although this was usually easy enough for any one autoradiograph, comparing data from one autoradiograph to the next, or worse still between different laboratories, could be considerably more difficult. Today, the replacement of minisatellite with microsatellite sequences in DNA profiling has largely overcome the binning problem, as will be discussed later. However, DNA profiling still remains subject to continuing statistical debates over the odds of chance matches; indeed, courtroom figures quoted by different “expert witnesses” can vary over a millionfold. Nevertheless, even the most cautious of these still place the odds of chance matches to be in the order of hundreds of thousands to one. Hence, although it may be inappropriate to consider it alone as positive proof, any court persuaded by defence lawyers to ignore forensic DNA evidence completely does so at its peril. Furthermore, it is vitally important to remember that the ability of forensic DNA evidence to exonerate the falsely accused is essentially absolute, and for this reason alone should be regarded as an indispensable tool.
Whereas at the time (before microsatellites) minisatellites were very useful to scientists for such things as paternity testing and forensic identification, when it came to linkage analysis minisatellites were similar to satellites, in that their uneven distribution throughout the genome made them relatively poor genetic markers. However, unlike satellites, which as mentioned previously, tend to cluster centromerically, minisatellites appear to prefer telomeric regions. Again, although there is no evidence to suggest that this restricted localisation has any functional relevance, minisatellites have been reported to act as “hot spots” for homologous recombination.15
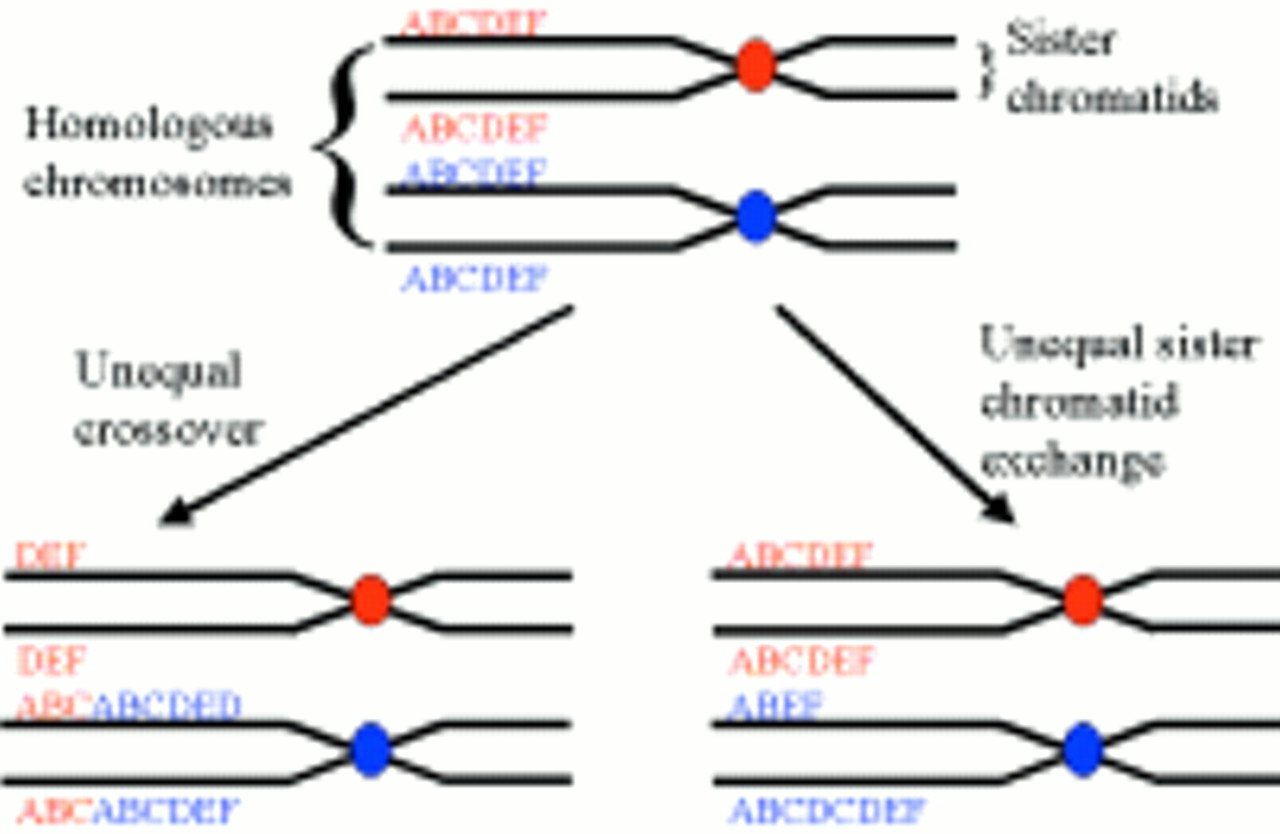
The precise genetic mechanisms that originated the huge expansions seen in satellite and minisatellite sequences are unclear. However, unequal crossover or unequal sister chromatid exchange probably accounts for the extreme variability often seen between individuals at these loci (fig 2).

Diagram illustrating the mechanisms of unequal crossover and unequal sister chromatid exchange, which are thought to be responsible for the extreme level of polymorphism seen in many satellite and minisatellite sequences.
Microsatellites
We have now reached the final stage in the hierarchy of tandemly repetitive sequences, namely microsatellites or STRs (short tandem repeats). These comprise simple mononucleotide to pentanucleotide repeats, varying from a few tens of bases up to typically one hundred or so, although some trinucleotide repeats make notable exceptions, as will be discussed later. They are spread more or less evenly throughout the entire human genome, with the total number of each decreasing as the size of the repeat unit increases. For example, mononucleotide repeats, especially A and T repeats, are believed to be present at around half a million loci (occupying about 10 Mb in total), whereas the number of pentanucleotide repeat loci is estimated at only a few thousand. It is probable that larger repeats are less common because of the way in which microsatellites are thought to arise. This involves a process that has been multiply referred to as “DNA slippage”, “polymerase slippage”, or “slipped strand mispairing”. In essence, this slippage is thought to occur within the complex of proteins that mediates DNA replication, as a consequence of mispairing (by one repeat unit or occasionally more) between the original template strand and the newly synthesised DNA strand. The resulting region of unpaired DNA is then forced to “loop out”. If this “loop” is on the new strand the net effect is addition of a repeat unit; if it is on the template strand it is removed by enzymes, and the net effect is loss of a repeat unit (fig 3). Larger order repeats would require either strand to slip further before the bases could pair correctly again, and would potentially explain why they are less common and often more stable. Other factors such as the number, location, and sequence of repeats are also likely to affect the rate and direction of slippage, although as yet no simple or universal rules have been found.16,17 However, it does appear that as expansions get larger there is a tendency for the loss of multiple repeats to predominate, hence providing a biological constraint to prevent unlimited growth.18 In addition to these factors, it must also be remembered that the rate at which slippage phenomena occur is not the same as a microsatellite's apparent mutation rate. This is because most loops resulting from slippage are repaired correctly by the cell's DNA mismatch repair system. Consequently, observed microsatellite mutation rates represent the net results of an interplay between slippage and mismatch repair.16 Indeed, geneticists may regard themselves as remarkably fortunate that the balance between these processes lies where it does. If the observed mutation rates were too low, microsatellites would be very rare and far less polymorphic; if they were too high, many would show changes from one generation to the next, preventing patterns of inheritance being investigated accurately. For example, were a child to possess alleles of a particular marker not present in either of its parents, we would infer that the child was adopted. However, this inference relies on the assumption that the odds of that marker undergoing mutation (to produce the alleles seen in the child) within a single generation are very low indeed. With human microsatellites it is almost always acceptable to make this assumption; nevertheless, one must remember that their net mutation rate remains about 10 000 times that for a single base change.19 Thus, in large scale projects, such as certain genome wide linkage studies (see below), inheritance patterns that can only be explained by germline mutation at a given microsatellite locus are to be expected occasionally (P Bennett, unpublished data, 1999).20

Strand slippage during DNA replication. (A) The extending strand slips backwards, resulting in the insertion of an extra repeat unit in the newly synthesised strand. (B) The extending strand slips forwards, resulting in the deletion of a repeat unit in the newly synthesised strand.
Before finally going on to consider how microsatellites can be useful to us as scientists or clinicians, it is interesting to consider how they can be of direct use to some of their host organisms. Certain types of bacteria are known to use microsatellite containing genes called “contingency genes”. These can confer survival advantages upon a subset of the bacterial population under changing environmental conditions. A specific example of this, which is also of pathogenic relevance to humans, is the use of contingency genes by Neisseria gonorrhoeae as a means of evading the immune system. These organisms possess about a dozen genes encoding outer membrane proteins, each of which contains very unstable pentanucleotide repeats. Because the amino acid sequences of proteins are encoded by three base pair units of DNA (codons), any slippage not divisible by this number will cause a frameshift and no protein will be expressed. Subsequent slippages that are divisible by three will of course restore expression. The result of this dynamic process is that only a few genes are switched on at any time; however, which ones these are continually varies from one bacterial generation to the next. This allows the bacterial population to vary its antigenic profile and hence evade the immune system.21 This reversible switching is called “phase variation”, and has been found in many disease causing bacteria.
Contingency genes appear to be confined to prokaryotes. However, allowing for the greater abundance of microsatellite sequences in eukaryotes, combined with their frequent localisation near to or even inside genes (especially trinucleotide repeats), it would be surprising if some did not play a biological role. Indeed, there is already evidence to suggest that glutamine rich domains and triplet repeat encoded polyglutamine stretches are integral components of many proteins involved in transcriptional regulation, from yeasts through to humans.22 This has led to the hypothesis that some microsatellites in higher organisms may be acting as “dimmer switches”, providing a finer level of control than the simple on/off of bacterial contingency genes.23
Whereas the biological actions of microsatellites in humans may be subtle, some of their biological consequences certainly are not. This is exemplified by the trinucleotide repeat associated diseases such as Huntington's, myotonic dystrophy, and certain types of spinocerebellar ataxia.24–27 Now known to include more than a dozen rare, dominant, and mainly neurological disorders, this group of diseases is characterised by the primary genetic cause being the expansion of a trinucleotide repeat far outside of its “normal” polymorphic range.17 Such repeats are usually inside the disease gene, wherein most encode runs of glutamine residues; others, which are outside, are close enough to disrupt its correct functioning. An interesting feature is that disease severity often appears to correlate with the extent of abnormal expansion. This, in conjunction with the fact that the longer these repeats become the more prone they seem to yet further expansion, may explain the anticipation (successive generations experiencing earlier onset or more severe symptoms) often seen in families afflicted by one of these trinucleotide repeat associated diseases. (Note: despite the name “trinucleotide repeat associated diseases” most trinucleotide repeats are not pathogenic and some might even serve useful functions as described above.)
Apart from the fact that trinucleotide repeats should be better tolerated within coding sequences (because three base pairs constitute a codon the reading frame is not disrupted), the genetic mechanisms responsible for pathogenic trinucleotide repeats being so different to other microsatellite sequences are not understood. However, clues as to why such mechanisms exist may come from the following interesting observation. To date, trinucleotide repeat associated diseases have only been identified in humans. This absence, even from such close relatives as the chimpanzee, combined with the fact that most trinucleotide repeat associated diseases affect neurological function, has led to the hypothesis that the presence of trinucleotide repeats within certain brain related genes may have increased adaptability and contributed to the evolution of brain function. This means that the trinucleotide repeat associated diseases could represent the genetic costs for those benefits that we now enjoy over other primates.19
So how are microsatellites used?
Microsatellites are almost ideal genetic markers. They are abundant, codominant, highly polymorphic and, very importantly, they are spread across the entire euchromatic part of the genome. Consequently, they have largely replaced minisatellites and now represent the markers of choice for many genetic applications. Indeed, so many people began using so many different microsatellite markers in so many different applications that a universally accepted system for naming and cataloguing them rapidly became essential. As a result, human microsatellite markers are now named in standard formats—for example, D12S324, where 12 is the chromosome on which the marker is located and 324 comprises a unique identifier. Information pertaining to each marker, such as cytogenetic location, heterozygosity, allele frequencies, and assay conditions, can be obtained via the internet (http://www.marshmed.org/genetics/ is a good site to start at). These internet websites are operated by large institutions involved in mapping/sequencing the human genome. The work is collaborative and information is exchanged regularly to ensure that the relevant globally accessible databases are kept up to date.
So how are microsatellite sequences assayed within the laboratory, and how are the data generated useful? This question is best answered by considering several common applications as examples.
In essence, most assays for microsatellites are similar to those described previously for minisatellites in that they involve the electrophoretic separation of DNA fragments according to size. However, unlike the large minisatellite containing DNA fragments generated by restriction enzyme digestion and separated in low resolution media such as agarose, microsatellite containing DNA fragments are usually small enough to be amplified using the polymerase chain reaction (PCR) and separated in high resolution media like polyacrylamide (some of the pathogenic triplet repeat expansions are exceptions). Because of this, alleles differing in size by only a single repeat unit can be resolved unambiguously. This fact alone has contributed towards solving the problems associated with the binning process mentioned previously, although as we shall see, additional technologies can provide even more improvement. Not all microsatellite based applications require allele binning—for example, diagnostic testing to determine which currently healthy family members are going to succumb to one of the trinucleotide repeat associated diseases. For some of these diseases, it may only be necessary to determine that the relevant repeat has undergone a gross expansion, whereas for others the threshold between getting the disease or not can be as little as a few repeat units. Loss of heterozygosity (frequently referred to simply as LOH), which is a technique used to identify the possible locations of tumour suppressor genes, is another application in which the precise determination of specific alleles at a given marker is not necessary. This is because the only comparisons being made are between DNA from normal and tumour tissue isolated from single heterozygous individuals. Any consistent loss of this somatic heterozygosity (regardless of allele) for a specific microsatellite marker is indicative of a DNA deletion within the tumour. In addition to the marker, this deletion will contain some part of the surrounding DNA, which may have contained a previously active copy of a tumour suppressor gene.
Of those applications that do require alleles to be binned, the most important one not yet discussed is linkage analysis. Not previously possible on a comprehensive genome wide scale, owing to the restricted chromosomal localisation of other types of markers, linkage analysis is a technique aimed at finding the approximate chromosomal location of any type of disease gene. This may be relatively easy for single gene disorders, with as little as a single large family sometimes being enough. For so called complex disorders that are not inherited in a simple Mendelian fashion, many hundreds of families might be required to obtain the statistical power needed to identify the number of much weaker disease susceptibility genes likely to be involved. Thus, such projects can rapidly become considerable undertakings generating huge amounts of data, the statistical analysis of which alone can be formidable. Nevertheless, the basic principles of genetic linkage analysis are relatively simple, relying essentially on two assumptions, namely: (1) that at a given locus each parental allele has an equal chance of being transmitted to a child, and (2) that as a consequence of the random recombinations that take place at meiosis, the smaller the genetic distance between two loci the greater the chance that they will be co-inherited. In short, microsatellite markers whose alleles are seen to be inherited preferentially by those family members with the disease are said to be linked, and should co-localise with the disease gene. On the other hand, the alleles of unlinked markers lying well away from the disease gene locus should be inherited equally between affected and unaffected family members. LOD scores which are statistics frequently quoted in linkage studies, provide a measure of the likelihood that any preferential inheritance seen for a given marker is the result of genuine linkage as opposed to simple chance.
For projects such as complex genetic linkage studies, there is already a considerable chance inherent in the methodology of generating false positives/negatives, hence it is essential that this is not increased by genotyping errors. As for paternity testing, and especially forensic DNA testing, it need not be explained how the consequences of genotyping errors could be dire indeed. Consequently, I shall conclude this review with a brief summary of the current state of the art for microsatellite genotyping.
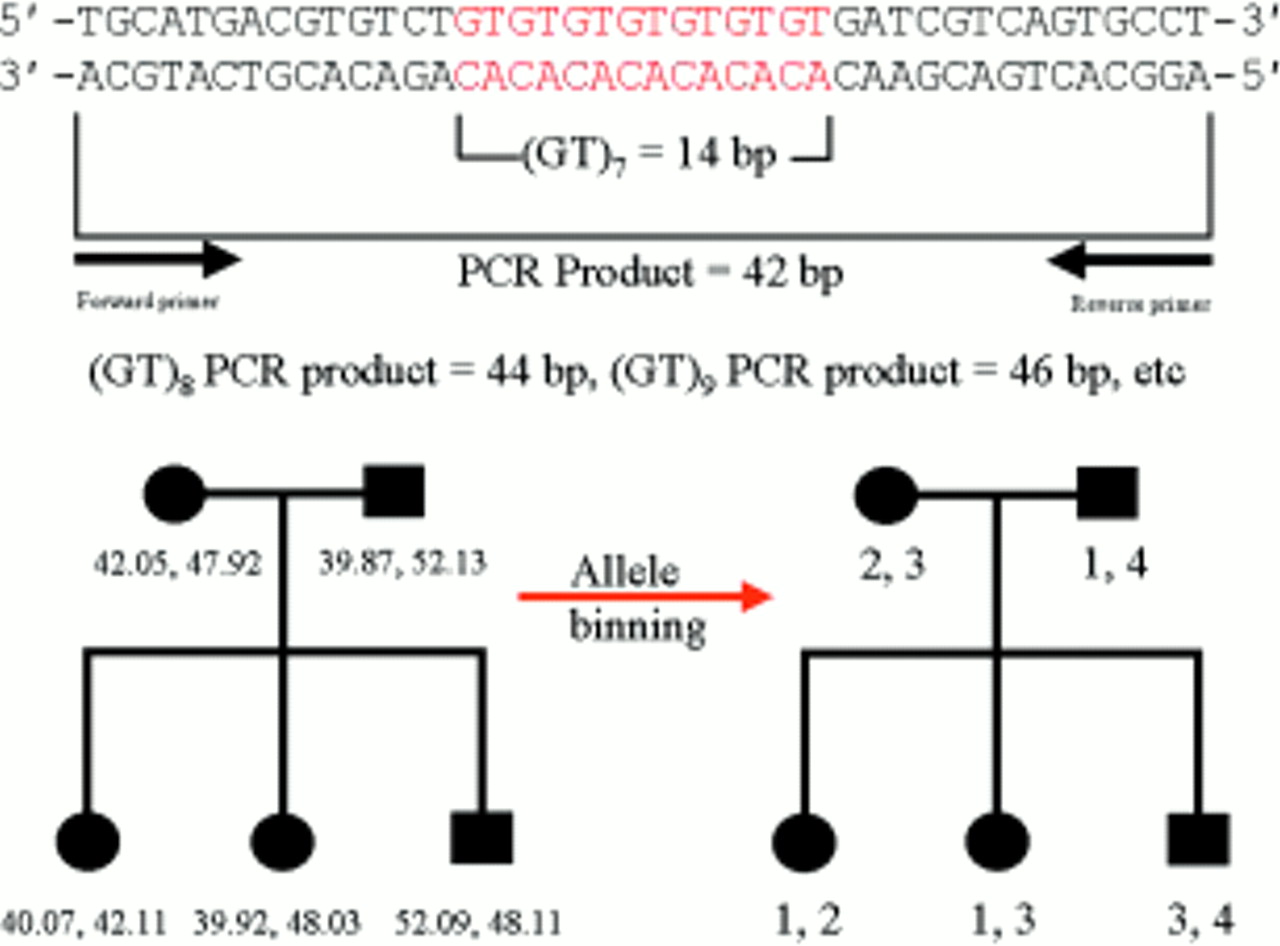
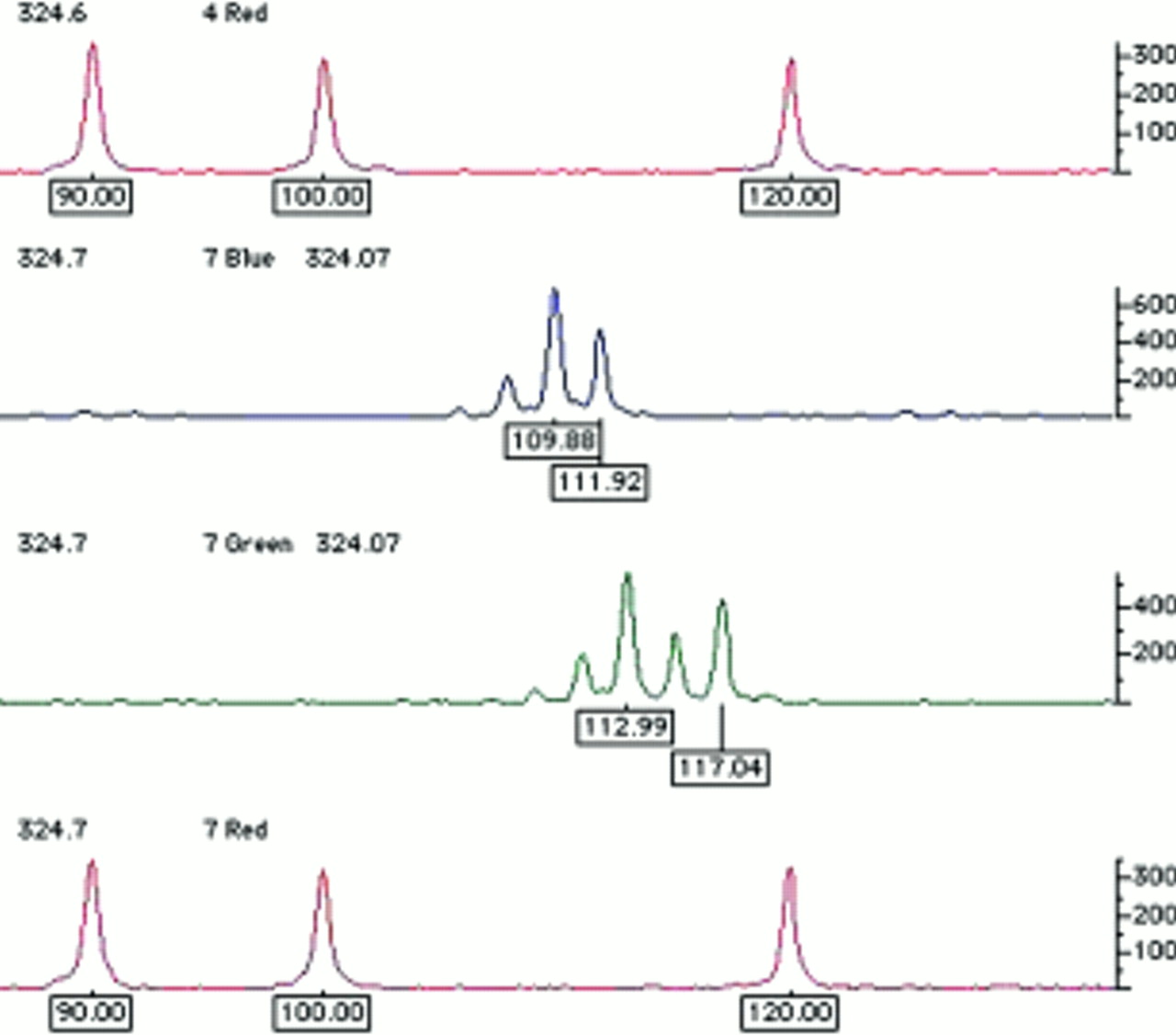
Traditionally, DNA fragments separated in polyacrylamide have been visualised by techniques such as autoradiography or silver staining. However, with these techniques all DNA fragments appear the same; thus, to avoid confusion between different fragments of similar size any reference standards used to estimate the size of the test fragment must be physically separated. At best, this will be in the adjacent lane on the gel; at worst, reference standards will be at the edges of the gel, with several dozen test samples between them. For several reasons, such gels rarely if ever run completely level; therefore, after visualisation one cannot simply place a ruler across and assume that aligned fragments are by definition the same size. This problem of lane to lane variation has now been resolved by the use of fluorescence technology. The reference size standards and PCR primers used to amplify the microsatellite sequences are covalently labelled with different fluorophores. These molecules all absorb light energy supplied by a laser, but then re-emit light at a different wavelength specific to each fluorophore. The different wavelengths can be interpreted by a computer and displayed as different colours in real time. As a result, it is now possible to analyse up to 20 or so microsatellites in a single gel lane, with their alleles' sizes calculated by computer, relative to a set of internal size standards labelled in a unique colour (fig 4). In short, it is only necessary to ensure that different fragments do not overlap in both size and colour; thus, as hardware and fluorophore technology progress it may be possible to analyse even larger numbers of markers simultaneously. After automated fragment sizing, allele calling is a simple task of relabelling fragments of the same size as the same allele. However, this is still a binning process. Because the computer's sizing algorithm will generate non-integer values (figs 4 and 5), maximum and minimum limits for inclusion in each particular allele bin must still be set. Nevertheless, these bins are now discrete and, because alleles do not fall in between them, they should never end up in the wrong bin altogether. For example, in fig 5, bin limits may be set at 39.8–40.2, 41.8–42.2, 47.8–48.2, and 51.8–52.2.

Examples of the raw output generated using semi-automated fluorescent technology to genotype microsatellite markers. Here, alleles at two different microsatellite loci that overlap in size range are resolved by labelling with different fluorophores (visualised in blue and green). Amplicons are labelled with their estimated size in base pairs, calculated automatically by comparison with the reference size standards shown in red. These size standards are premixed with the test sample and electrophoresed simultaneously in the same gel lane, automatically controlling for lane to lane variation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic illustrating a hypothetical microsatellite sequence after PCR amplification. After automated amplicon sizing, the process of allele binning is shown to illustrate the way in which the Mendelian inheritance of microsatellite marker alleles can easily be followed. For simplicity, the microsatellite containing polymerase chain reaction amplicon shown is very small, typically amplicons would be at least twice this size.
The technology described above is now standard in many laboratories, although in due course it may change, and in certain applications microsatellites may themselves be superseded as the markers of choice. For example, there is debate that for linkage analysis single nucleotide polymorphisms may possess certain advantages over microsatellites, and could ultimately replace them. However, for areas such as forensic work, the relative ease of assay, combined with the high degree of polymorphism available at a single locus, mean that routine high throughput typing of human microsatellites is likely to remain with us for some time.