Article Text

Abstract
Background/Aim—The pathogenetic relation between liver cell dysplasia and hepatocellular carcinoma (HCC) is poorly understood. The aim of this study was to determine whether there is a genetic link between liver cell dysplasia and HCC that could support the role of dysplasia as a tumour precursor lesion.
Methods—Microdissection from paraffin wax embedded sections and degenerate oligonucleotide primed polymerase chain reaction (DOP-PCR) were combined to analyse chromosomal imbalances by comparative genomic hybridisation (CGH) in nine HCCs and nodules containing liver cell dysplasia and cirrhosis adjacent to the tumours. Seven cases of large cell changes (LCC) and three cases of small cell changes (SCC) were analysed. The genetic abnormalities detected in liver cell dysplasia were then compared with those present in the corresponding HCC.
Results—No abnormalities were detected in LCC and cirrhotic nodules, arguing against the preneoplasic nature of these cell foci. In contrast, a subset of chromosomal alterations present in HCCs was found in the adjacent SCC.
Conclusions—These findings support the preneoplastic status of SCC in human hepatocarcinogenesis.
- comparative genomic hybridisation
- hepatocellular carcinoma
- liver cell dysplasia
Statistics from Altmetric.com
Hepatocellular carcinoma (HCC) is one of the most common human malignancies, yet the molecular mechanisms involved in its development and the preneoplastic cellular component still remain obscure. A characterisation of the genetic events that accompany the progression of precursor lesions towards fully malignant tumours may be useful for early detection, in addition to preventive treatment. In liver, two types of lesion are usually recognised as preneoplastic. The first consists of nodular lesions called dysplastic nodules,1 and several lines of evidence suggest that they pose a risk for malignant transformation.2–5 However, opinions on the histological severity attributed to such lesions vary widely among pathologists.6 The second type of preneoplastic lesion, referred to as liver cell dysplasia, is defined by the presence of atypical hepatocytes. According to most authors, liver cell dysplasia can be divided into two subtypes: large cell changes (LCC) and small cell changes (SCC).7, 8 The frequent association of such dysplastic foci with HCC arising in cirrhotic livers suggests a preneoplastic role for liver cell dysplasia in the sequence driving cirrhosis towards HCC.9–11 Although two large prospective studies have suggested that LCC is the most important risk factor for HCC,12, 13 its premalignant nature is still controversial.13–16 To date, there are no genetic reports supporting a direct transition from liver cell dysplasia to HCC.
Recently, the genetic analysis of morphologically defined cell foci from tissue sections has become feasible through the use of microdissection approaches. Comparative genomic hybridisation (CGH) is a powerful molecular cytogenetics tool that permits whole genome screening for quantitative genomic abnormalities without the requirement for an initial tissue culture step. In our study, we combined microdissection and CGH to identify chromosomal aberrations in cirrhosis, liver cell dysplasia, and HCC in nine liver resections. Our aim was to identify a possible genetic link supporting the role of liver cell dysplasia as a precursor lesion of HCC.
Materials and methods
PATIENTS AND TISSUE SAMPLES
Formalin fixed liver tissue specimens were obtained from nine patients who underwent complete resection for HCC in the surgical department of Beaujon Hospital (Clichy, France). All of the tumours developed on liver cirrhosis and contained dysplastic foci. By definition, these dysplastic foci are not macroscopically recognisable and are smaller than 1 mm in diameter. Such a pattern distinguished them from the dysplastic nodules.1 The aetiology of cirrhosis was viral in five patients (four hepatitis B virus (HBV), one hepatitis C virus (HCV)), alcoholic in three, and haemochromatosic in one. According to Edmonson and Steiner's criteria,17 six HCCs were grade II (moderately differentiated) and three were grade III (poorly differentiated). As described by Anthony et al,7 LCC was identified in seven patients as hepatocytes larger than normal with enlarged, hyperchromatic, often pleiomorphic nuclei and prominent nucleoli. LCC were composed either of few scattered cells within a cirrhotic nodule or, alternatively, as big foci occupying a whole nodule.8 SCC was identified by the criteria of Watanabe et al as small hepatocytes with nuclear atypia forming hepatic plates two to three cells in thickness and arranged in compact areas.8 The reticulin framework was preserved and architectural atypia was not sufficient to allow a definite diagnosis of HCC. All these dysplastic changes were encountered on systematic specimens performed in non-tumoral liver. No macronodules were present and no reactive fibrosis at the periphery of these dysplastic foci was observed. In summary, the three alcoholic cases were associated with LCC (E1, E4, E6), the haemochromatosic patient with both LCC and SCC (E2), the HCV infected patient with SCC (E8), three of the HBV infected patients with LCC (E5, E7, E9), and one HBV infected patient with SCC (E3).
MICRODISSECTION AND DNA EXTRACTION
For each case, appropriate tissue blocks were selected and serial sections, 7 μm in thickness, were dewaxed and stained either by haematoxylin and eosin for histological analysis or by Giemsa for microdissection. To avoid contamination during microdissection, two slides were selected for each case: one containing the tumour and the second both cirrhosis and dysplastic foci. Areas of interest, containing approximately 30 cells, were microdissected with a sterile extended pipette of 60 μm in diameter under an inverted microscope using either ×40 or ×100 magnification. Typical examples of LCC and SCC microdissected regions are shown in fig 1. Microdissected cells were collected in 5 μl of a solution (10mM Tris/HCl, pH 7.5, 10mM NaCl, 0.1% sodium dodecyl sulphate) containing 0.5 mg/ml proteinase K. Samples were incubated for several hours in a moist chamber at 65°C, followed by boiling for 10 minutes to inactivate the proteinase K.

Serial tissue sections of samples with (A, B) small cell changes (SCC) and (D, E) large cell changes (LCC) after haematoxylin and eosin staining (A, D) and the corresponding area after removal of the dysplasic cells of sections stained by Giemsa (B, E). (C) Higher magnification of an SCC sample (×200) and (F) an LCC sample (×100); the dysplastic areas are delimited by arrows.
For each cellular subtype (cirrhotic, dysplastic foci, and HCC) at least two distinct samples were microdissected. Genomic DNA extracted from 30 peripheral blood lymphocytes of healthy donors was used as a source of normal DNA. Lymphocytes were fixed three times in a methanol/acetic acid solution (3/1), and stained by Giemsa.
DEGENERATE OLIGONUCLEOTIDE POLYMERASE CHAIN REACTION (DOP-PCR)
DOP-PCR was performed following two steps on a thermocycler (PTC-100, MJ Research, Watertown, Massachusetts, USA). Eight initial cycles (preamplification step) were carried out in a 6 μl reaction volume (200μM of each dNTP, 0.6× Sequenase buffer (Amersham, Cleveland, Ohio, USA), 5μM UN1 primer (5′-CCG ACT CGA GNN NNN NAT GTG G-3′)) with one minute at 96°C, 2.20 minutes at 25°C, and two minutes at 37°C, with the addition of 0.4 U of Sequenase version 2.0 (Amersham) at each of the 25°C steps. After this low stringency preamplification, 34 cycles were performed in a 50 μl reaction volume (200mM dNTPs, 1mM UN1 primer, 2.5mM MgCl2, 1× Stoffel buffer (Perkin-Elmer, Branchbury, New Jersey, USA), and 1 U AmpliTaq DNA polymerase, Stoffel fragment (Perkin-Elmer)), with one minute at 94°C, one minute at 56°C, two minutes at 72°C, and a final extension of 10 minutes at 72°C.
DNA LABELLING
The DOP-PCR amplification products (4 μl) were labelled by a third amplification step using 0.8 μl of fluorescein 12-dUTP and 0.8 μl of fluorescein 12-dCTP for test DNA or Texas red 5-dUTP and Texas red 5-dCTP for normal DNA in a 20 μl reaction volume (1× Replitherm buffer (Biozym, Amelyn, Germany), 2.5mM MgCl2, 2.5mM dATP and dGTP, 1.25mM dTTP and dCTP, 2mM UN1 primer, and 0.6 U AmpliTaq DNA polymerase), with three minutes at 95°C followed by 25 cycles of one minute at 94°C, one minute at 56°C, two minutes at 72°C, and a final extension of five minutes at 72°C.
COMPARATIVE GENOMIC HYBRIDISATION (CGH)
Metaphase cells for CGH experiments were prepared from phytohaemagglutinin stimulated lymphocytes of healthy men. Lymphocyte cultures were synchronised by the thymidine method.18 The slides were hybridised for 72 hours with 20 μl of DOP-PCR labelled DNA in the presence of 50 μl Cot-1 DNA and 50 μg of sonicated salmon sperm DNA. After hybridisation, the slides were washed for two minutes in 0.4× saline sodium citrate (SSC) at 74°C and one minute in 2× SSC/0.1% NP40 at room temperature. DNA was counterstained with 0.2mM DAPI (4,5-diamino-2-phenylindole) in antifade solution.
DATA ACQUISITION AND PROCESSING
For each hybridisation, 10 metaphase cells were analysed using a Zeiss axioskop fluorescence microscope (Zeiss, Oberhochen, Federal Republic of Germany) and a Quips fish digital analysis system (Vysis, Illinois, USA) composed of a computer driven cooled CCD colour camera and Quips-XL software automated analysis software (Vysis). Relative changes in the copy number of DNA sequences were analysed using an adapted digital image analysis system. Chromosome regions were interpreted as over-represented if the corresponding colour ratio was higher than 1.25 and as under-represented if the ratio was lower than 0.75.
The amounts of DOP-PCR product and labelled DNA used in the experiments were normalised by the comparative analysis of a classic CGH, with DNA obtained from a frozen tumour, and a DOP-CGH with DNA from the corresponding microdissected region of the same tumour. The DOP-PCR product of each microdissection was hybridised at least twice and associated with normal DNA from two different healthy donors.
Results
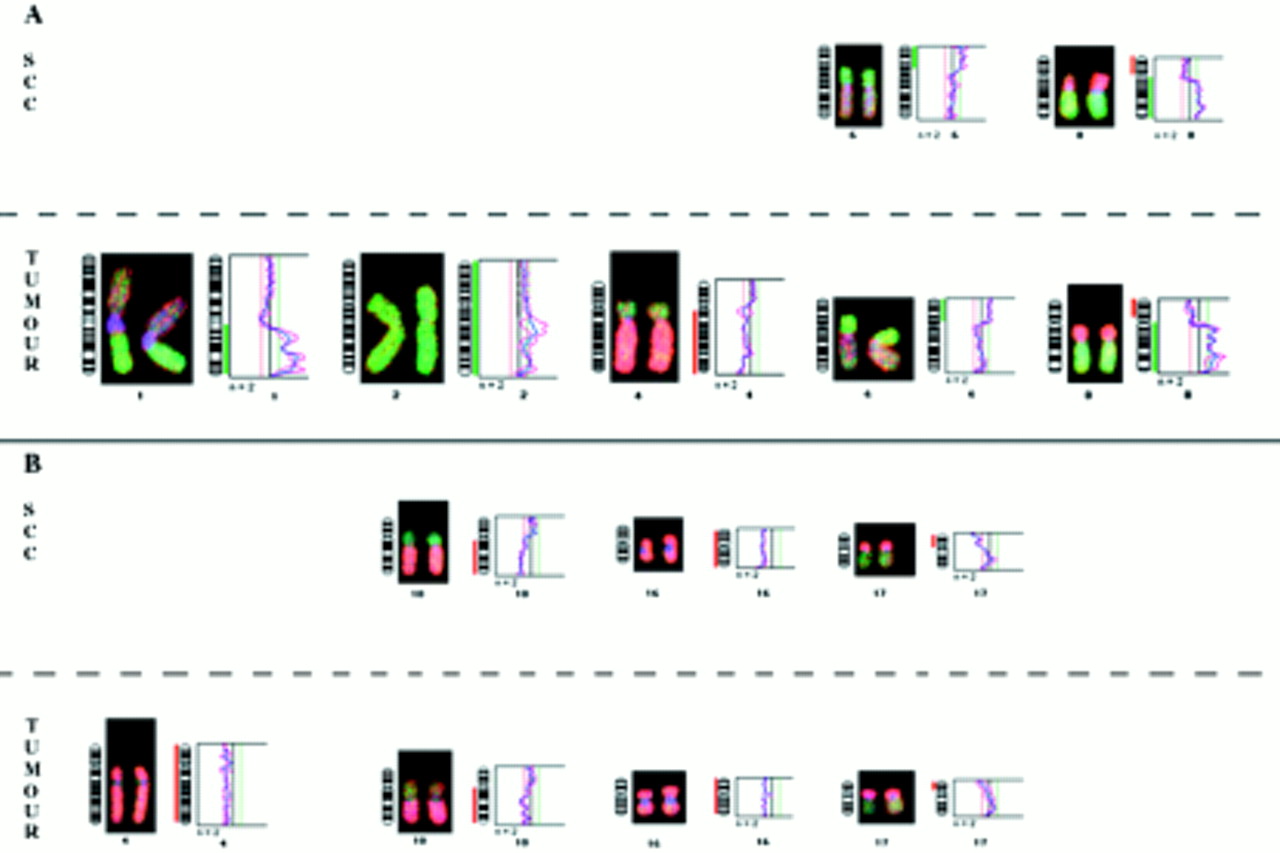
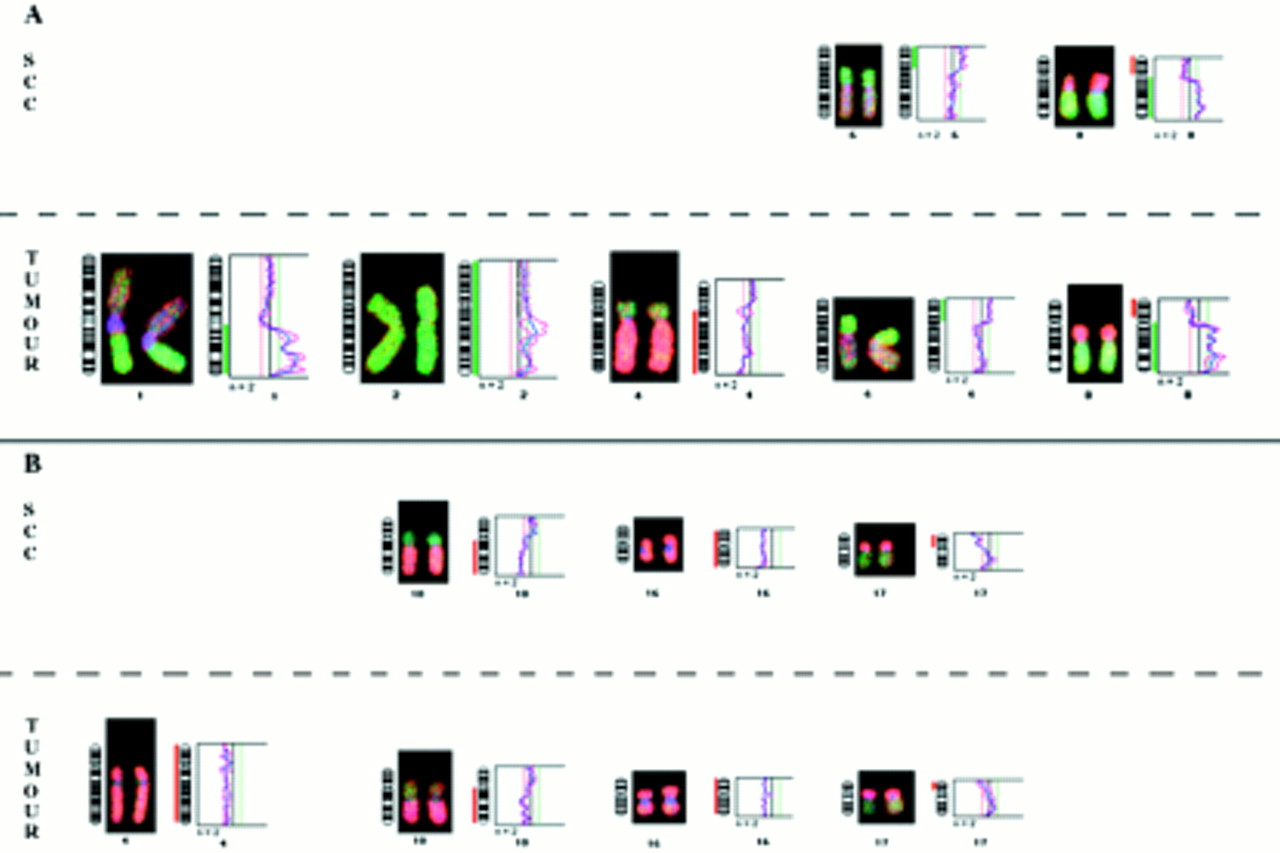
No abnormality was detected in the nine benign cirrhotic nodules analysed. Four of the nine HCCs and the corresponding dysplasias revealed no abnormalities by DOP-CGH. As shown in table 1, gains of chromosome arms 1q (four of nine) and 8q (three of nine) and loss of 8p (three of nine) were the most frequent alterations seen in the tumours (for example, see fig 2). These results agree with previous data implicating these chromosomal imbalances as major abnormalities associated with HCC.19, 20 None of the LCC found with these tumours (E2, E7, and E9) revealed abnormalities. In contrast, two of the three SCC (E3 and E8 but not E2) exhibited a subset of the abnormalities found in the tumours. In the SCC from the HBV infected patient (E3), the loss of 8p and gains of 6p and 8q were detected (table 1). Such genetic changes frequently found in full blown HCC are probably early events in tumour development. In contrast, other alterations highly prevalent in HCC, such as 1q gains or 4q losses, were not found in SCC. In the samples from the HCV infected patient (E8), three of the four abnormalities found in the tumour (losses of 10q, 16, and 17p) were also seen in the SCC.
Copy number changes found in informative small cell changes (SSC), large cell changes (LCC), and tumours (T)
{kind=link}
{kind=link}
Chromosomal abnormality pattern found in small cell changes (SCC) and adjacent tumour of patients E3 (A) and E8 (B).
Discussion
As with many other carcinomas, the development of HCC is characterised by an accumulation of genetic alterations. We and others have previously reported, either by molecular genetic studies21, 22 or by CGH,20 the frequent loss of chromosomes 1p (30–40%), 4q (40–70%), 8p (30–65%), 16q (30–50%), and 17p (30–50%) and gains of chromosomes 1q (60–80%), 6q (30–40%), 8q (60–70%), and 17q (30%). In many tissues, cell dysplasia is recognised as a precancerous lesion. However, the preneoplastic nature of liver cell dysplasia remains controversial because no genetic pathway has been established to link liver cell dysplasia to HCC.23, 24
Some authors consider LCC to be a regenerative or degenerative condition,16, 25 whereas others have shown by prospective studies that LCC is an important risk factor for HCC.12, 13 Because of the lack of histological continuity between LCC and HCC, the comparison of their genetic abnormalities could be useful in providing evidence for the possible preneoplastic status of LCC. We have previously shown by interphase cytogenetic analysis that LCC are composed of polyploid cells.26 In our present study, none of the microdissected LCC foci revealed chromosomal imbalances even when the adjacent HCC displayed such alterations. Our results do not support a direct relation between LCC and HCC. The lack of relevant genetic abnormalities in LCC, combined with their multiploidy and low proliferation rates, suggests that the appearance of such cells could result from impaired DNA replication in hepatocytes, as previously suggested by Altmann.27 Thus, LCC might be a consequence of chronic injury in a subpopulation of terminally differentiated or “senile” hepatocytes of limited proliferative capacity,14, 15 and is therefore more likely to be a pericancerous marker, rather than a true precancerous lesion.28 Similarly, cirrhotic nodules, which could also be considered as precancerous lesions, failed to exhibit chromosome segment gains or losses in the nine specimens analysed, confirming previous observations.20, 29
To date, there are no uniform criteria for defining SCC, a situation reflected by the broad variations in its detection by pathologists examining liver cirrhosis. In our study we defined SCC as a group of cells showing an “expansive pattern” with smaller amounts of cytoplasm than normal hepatocytes and an increased nuclear/cytoplasmic ratio (fig 1). However, these cytological and structural atypia are not sufficient to allow a definite diagnosis of well differentiated HCC (or grade I according to the Edmonson and Steiner criteria).17 Furthermore, such foci did not correspond to intrahepatic metastasis of the adjacent HCC, because in all cases with SCC the latter showed a moderately differentiated pattern. SCC has been reported to be a preneoplastic change because of its phenotypic similarities with well differentiated HCC and its higher incidence in cirrhotic livers bearing HCC.8 To date, however, no studies have attempted to identify genetic changes occurring in such cells. In two of the three foci of SCC analysed, we detected chromosomal alterations very similar to those present in the adjacent HCC. These results suggest that SCC is a preneoplastic lesion. In SCC, the absence of 1q amplification and 4q loss (the most frequent abormalities found in HCCs) suggests that these abnormalities are associated with tumour progression. Furthermore, loss of 8p and gain of 8q found in a SCC may correspond to earlier events.30 In contrast, losses of 16q and 17p, reported to be linked to HBV infection,31 are present in SCC, suggesting an association of these abnormalities with tumour initiation. The loss of the 10q arm is a rare event in HCCs of non-B, non-C, and HBV infected patients, but is seen frequently in HCCs associated with HCV infection.32 Loss of 10q detected in the HCV associated SCC suggests an early involvement of this abnormality in HCV induced HCC.
Our study demonstrates that the combined approach of microdissection, DOP-PCR, and CGH is suitable to identify early genetic change in liver cell dysplasia. A recent report from Libbrecht et al showed that SCC and the putative liver progenitor cells, but not LCC, exhibit the same immunohistochemical phenotype.33 In addition, the presence of such progenitor cells in more than 60% of SCCs suggests that these cells can give rise to foci of SCC. Taken together, these results may lead to a consistent model of human hepatocarcinogenesis. Further molecular analysis of the putative progenitor cells contained in SCC should hopefully establish a genetic link between an individual cell type and HCC.
Acknowledgments
This work was supported by a grant from Association pour la Recherche sur le Cancer (ARC), la Ligue Nationale contre le Cancer, and the Société Nationale Française de Gastroentérologie. PP was supported by a grant from the Fondation Pasteur-Weizman. We warmly thank J S Seeler for his careful reading of the manuscript.