Article Text

Abstract
Background: Formalin fixed and paraffin wax embedded tissues of necropsy origin are an important source for molecular analysis especially in rare diseases, neuropathology, or molecular epidemiology studies. Because of DNA degradation, only short sequences can be amplified from this type of tissue, very often less than 100 bases. This poses problems because studies on polymorphism and mutations occurring in large genes often require the analysis of long sequences.
Methods: The development of a simple treatment to obtain longer fragments of DNA for the analysis of archival postmortem paraffin wax embedded tissues.
Results: It was possible to amplify longer sequences ranging up to 300 bases from postmortem tissues, with no modification to the usual DNA extraction procedures. To obtain longer stretches of DNA, a pre-PCR restoration treatment was required, by filling single strand breaks, followed by a vigorous denaturation step.
Conclusions: The development of this simple treatment allowed the analysis of longer fragments of DNA obtained from archival postmortem paraffin wax embedded tissues.
- formalin fixed paraffin embedded tissues
- polymerase chain reaction
- DNA degradation
- postmortem tissues
- ApoE, apolipoprotein E
- PCR, polymerase chain reaction
Statistics from Altmetric.com
Paraffin wax embedded tissues are an extraordinary source for DNA molecular studies because of the availability of large pathology archives of tissues related to clinical cases in almost all hospital pathology departments. In addition to biopsy and surgical paraffin wax embedded tissues, postmortem tissues are an important resource, especially for rare diseases, neuropathology studies,1 or molecular epidemiology studies,2 because both pathological and normal tissues can be analysed. The major difficulty in using these tissues is the degradation of nucleic acids, which is more extensive than in paraffin wax embedded tissues from biopsies or surgical specimens.
“The average fragment length of DNA is 300–400 bases in biopsy tissues, but much shorter in postmortem paraffin wax embedded tissues”
Tissue processing is a source of great variability, and in routine clinical laboratories the procedure is not completely standardised. This variability results from the type of fixative solution used, the storage conditions (especially formaldehyde solutions), and the suppliers. Some authors have reported that tissue storage in formaldehyde solution for more than one week might damage nucleic acids, because fixation in formaldehyde induces extensive crosslinking of tissue proteins, resulting in nucleic acid fragmentation.3 Tissues should be fixed in a buffered formaldehyde solution (the pH must be in a physiological range), in the dark for 24 hours before paraffin wax embedding. Postmortem tissues are usually fixed for a longer period of time. More extensive degradation of nucleic acids is often found in archival tissues older than 20 years because non-buffered formaldehyde solution was often used in the past. A non-buffered formaldehyde solution oxidises to formic acid and an acidic environment is the main reason for DNA degradation. However, DNA is relatively stable in mildly acidic solutions, but at around pH 4 the β glycosidic bonds in the purine bases are hydrolysed. Protonation of purine bases (N7 of guanine and N3 of adenine) occurs in this acidic environment. Protonated purines are easily cleaved, hence the hydrolysis. Once this happens, the depurinated sugar can easily isomerise into the open chain form, and in this form the depurinated or apurinic DNA is susceptible to cleavage by hydroxyl ions.4
The average fragment length of DNA is 300–400 bases in biopsy tissues, but much shorter in postmortem paraffin wax embedded tissues.3, 5 In postmortem tissues, fixed in non-buffered formalin, DNA fragments longer than 90 bp cannot be amplified. DNA degradation in paraffin wax embedded tissues is usually connected with single strand breaks.6
As a consequence of DNA degradation, only very short sequences can be amplified in postmortem tissues, but longer fragments are necessary for many analyses. A partial restoration and reconstruction of DNA length in these cases is possible. As previously reported, we assumed that a partial reconstruction of DNA could be done in postmortem tissues by filling in the breaks in the DNA.7 Here, we show that it is possible to analyse human postmortem paraffin wax embedded tissues amplifying a 287 bp sequence of apolipoprotein E (ApoE) and 291 bp of the prealbumin gene (TTR).
MATERIALS AND METHODS
Twelve hepatic tissues, three from biopsies and nine from necropsies, were obtained from the department of pathology of the University of Trieste, Italy. The necropsies were carried out between 1972 and 1999. DNA from a human hepatoma cell line (HEPG2) was used as a control.
Extraction
DNA was extracted from 6 μm sections of paraffin wax embedded tissues. Ten sections were cut with standard microtomes for every paraffin wax block. To prevent cross contamination between the samples, the blade was shifted after each block. As previously reported,8 paraffin wax was removed by two xylene washes, followed by two ethanol washes—100% and 70%. After air drying, the tissue pellets were digested overnight at 45°C with proteinase K (0.5 μg/ml) in 50mM Tris/HCl, 1mM EDTA, and 0.5% Tween 20. To purify DNA from proteinase K and proteolysis residues an extraction with phenol-Tris/chloroform (1/1) was performed. The final sample of DNA was obtained by precipitation with ethanol using glycogen as the carrier. After washing the DNA pellet with 70% ethanol, it was air dried, resuspended in TE buffer, and quantified using an ultraviolet spectrophotometer.
Pre-PCR DNA restoration treatment
The DNA restoration process7 comprises a pre-polymerase chain reaction (PCR) treatment. DNA samples were incubated for one hour at 55°C in 100 μl of solution containing 10mM Tris/HCl (pH 8.3), 1.5mM MgCl2, 2% Triton X-100, and 200 μM of each dNTP. After this incubation, 1 U Taq DNA polymerase (Amersham) was added and DNA polymerisation was performed at 72°C for 20 minutes. The treated samples were then stored at −20°C until processed. This restoration method is based on the fact that DNA degradation is connected to random single strands breaks. The polymerase reaction restores the nicks after DNA rehybridisation, using the other strand as the template. Because of the random position of the nicks, the fragment reconstruction gives sequences long enough for successful amplification.
Denaturation step
The denaturation step, before PCR amplification, was performed on the pre-PCR treated samples, using 10 μl aliquots, which were denatured at 95°C for five minutes and then immediately chilled on ice. After this step the samples were added to the PCR solution.
Ligation step
To evaluate the possibility of further elongation, the pre-PCR treated samples were also subjected to a ligation step after renaturation. Ligation was performed in a 30 μl final volume. Every reaction included 10 μl of pre-PCR treated DNA, 6mM Tris/HCl (pH 7.5), 6mM MgCl2, 5mM NaCl, 0.1 μg/μl bovine serum albumin, 7mM β mercaptoethanol, 0.1mM ATP, 2mM ditiothreitol, 1mM spermidine, and 8 U T4 DNA ligase (Clontech, Palo Alto, California, USA). The reaction was allowed to proceed overnight at 16°C.
Amplification
The following primers were used for the PCR analysis:
-
ApoE (forward): 5′-AAG GAG TTG AAG GCC TAC AAA T-3′.
-
ApoE (reverse): 5′-GGC CTG GTA CAC TGC CAG-3′.
-
TTR1 (forward): 5′-CAG CAG GTT TGC AGT CAG AT-3′.
-
TTR1 (reverse): 5′-GGT ACC CTT GCC CTA GTA AT-3′.
-
TTR4 (forward): 5′-TGG TGG AAA TGG ATC TGT CTG-3′.
-
TTR4 (reverse): 5′-TGG AAG GGA CAA TAA GGG AAT-3′.
PCR was performed in a 50 μl final volume under standard conditions. Each reaction included 250 ng of DNA, 15 pmoles of each primer, 50mM KCl, 1.5mM MgCl2, 10mM Tris/HCl (pH 9 at room temperature), 200 μM of each dNTP, and 1 U Taq polymerase (Amersham Biosciences, Uppsala, Sweden). For Apo E analysis, PCR was carried out using the following amplification programme: denaturation at 95°C for three minutes, five cycles of 95°C for one minute, 60°C for one minute, and 72°C for one minute, followed by 45 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. The length of the Apo E fragment was 287 bases.
To establish the maximum length to be amplified in postmortem tissue with this method two sequences from the human prealbumin gene (GeneBank M11844) were amplified. In this experiment, the analysis was extended to a total of nine postmortem liver tissues instead of the four used previously to set up the method. The primers used selected a region of 291 bases (496–787) located on exon 1 and a region of 339 bases (7235–7574) located on exon 49 of the prealbumin gene. PCR analysis was performed as reported previously for the Apo E gene.
RESULTS AND DISCUSSION
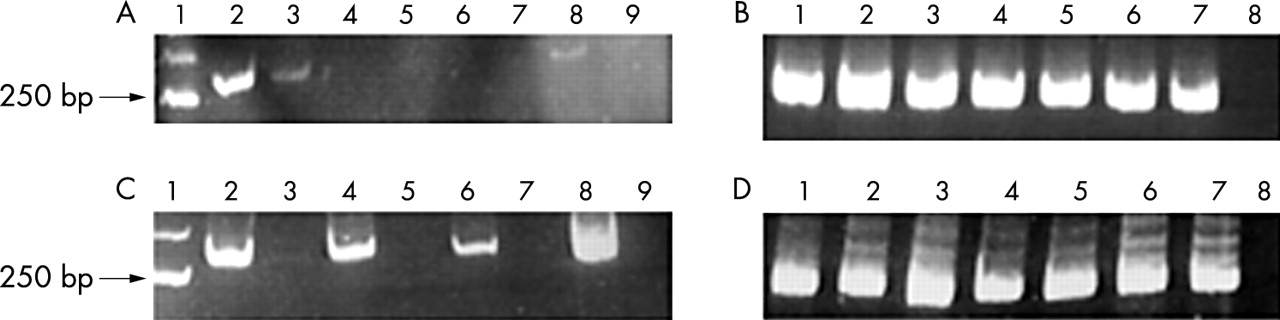
DNA samples of both postmortem and biopsy origin, with and without the pre-PCR restoration step, were amplified (table 1). As shown in fig 1A, no amplification of Apo E was detected in postmortem tissues, both in treated and untreated samples. However, for biopsy tissues (fig 1B), the PCR reaction was always successful, even in untreated samples. To obtain PCR amplification with restored DNA in postmortem tissues a denaturation step at 95°C for five minutes was performed after the pre-PCR restoration and the samples were immediately chilled on ice and then added to the PCR solution. As shown in fig 1C, amplification was positive only in restored and denatured postmortem DNA. Our hypothesis is that the negative results obtained for postmortem tissues, even after the restoration of the single strand breaks, are not caused by the failure of the DNA restoration procedure. Instead, these negative results could be a direct consequence of the restoration itself, where the double stranded DNA is strongly linked, with a more stable secondary structure. The fact that PCR amplification was obtained in formalin fixed, paraffin wax embedded biopsy specimens even without the restoration procedure shows that more extensive DNA degradation is confined to postmortem tissues. This problem can be overcome with a restoration/denaturation procedure.
PCR amplification of postmortem and biopsy DNA with and without the restoration and denaturation steps
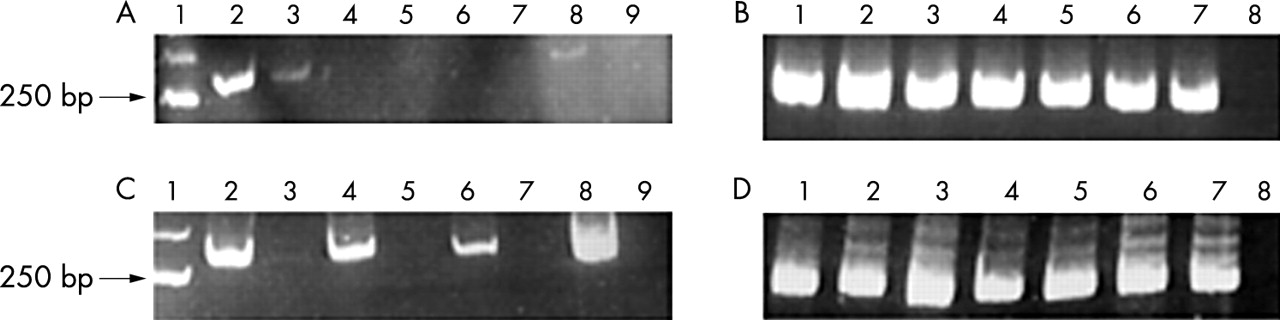
PCR products for apolipoprotein E (ApoE) analysis in paraffin wax embedded tissues of necropsy and biopsy origin. (A) Postmortem DNA analysis without the denaturation step. Molecular size marker (lane 1), postmortem DNA cases with the restoration step (lanes 2, 4, 6, and 8), and the same postmortem DNA cases without the restoration step (lanes 3, 5, 7, and 9). (B) Biopsy DNA analysis without the denaturation step. Positive control (lane 1), biopsy DNAs with the restoration step (lanes 2, 4, and 6), the same biopsy DNAs without the restoration step (lanes 3, 5, and 7), and the negative control (lane 8). (C) Postmortem sample analysis with the denaturation step. Molecular size marker (lane 1), postmortem samples with the restoration and denaturation steps (lanes 2, 4, 6, and 8), the same postmortem samples without the restoration step (lanes 3, 5, 7, and 9). (D) Biopsy sample analysis with the denaturation step. Positive control (lane 1), biopsy samples with the restoration and denaturation steps (lanes 2, 4, and 6), the same samples without the restoration step (lanes 3, 5, and 7), and the negative control (lane 8).
Take home messages
-
We have developed a method for amplifying longer DNA sequences, ranging up to 300 bases, from postmortem formalin fixed and paraffin wax embedded tissues, with no modification to the usual DNA extraction procedures
-
Before the polymerase chain reaction was carried out we used a restoration treatment, filling in single strand breaks, which was followed by a vigorous denaturation step
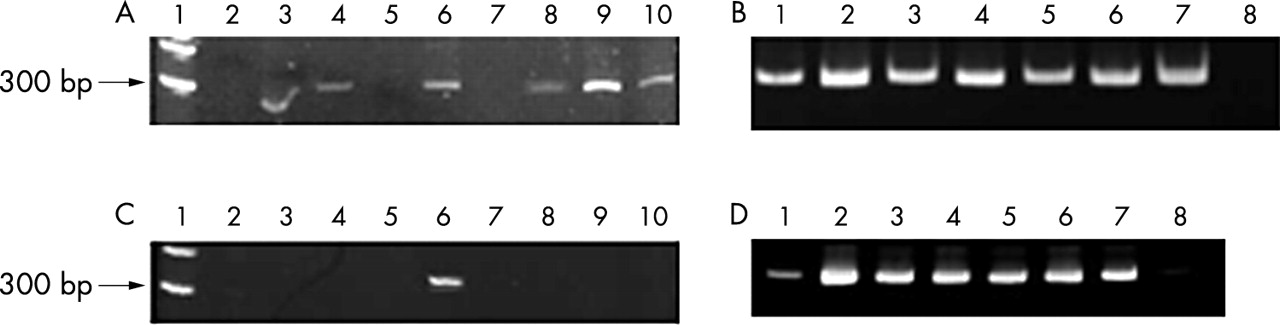
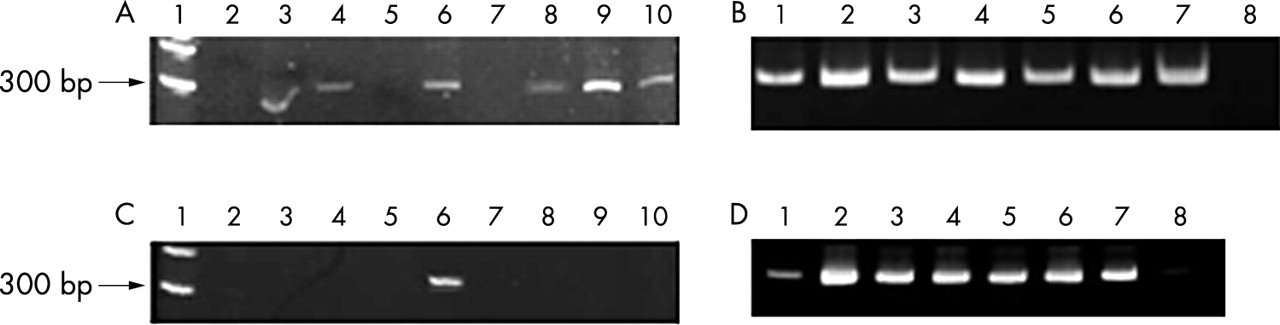
To evaluate the maximum length of amplification, two sequences of the prealbumin gene were also analysed. One PCR fragment of 291 bases in length and a second longer set of primers with an amplicon of 339 bases were selected. As shown in fig 2, 291 base fragments were amplified in six of the nine postmortem tissues, whereas for biopsy samples positive amplification was seen both in treated and untreated specimens. The amplification of the 339 base fragment was detected in only one postmortem tissue and again in all biopsy tissues. These results confirm the hypothesis that extensive DNA degradation occurs mostly in postmortem tissues and that with DNA restoration procedures it is possible to amplify fragments of about 300 bases.
{kind=link}
{kind=link}
PCR products in paraffin wax embedded tissues of necropsy and biopsy origin. (A) Postmortem sample analysis for the TTR gene (291 bases). Size marker (lane 1), postmortem samples with the treatment and denaturation steps (lanes 2–10). (B) Biopsy sample analysis for the TTR gene (291 bases). Biopsy samples with the treatment and denaturation steps (lanes 2, 4, and 6), the same biopsy samples without treatment (lanes 1, 3, and 5), the positive control (lane 7), and the negative control (lane 8). (C) Postmortem sample analysis for the TTR gene (339 bases). Molecular size marker (lane 1), postmortem samples with the treatment and denaturation steps (lanes 2–10). (D) Biopsy sample analysis for TTR gene (339 bases). Biopsy samples with the treatment and denaturation steps (lanes 2, 4, and 6), the same biopsy samples without treatment (lanes 1, 3, and 5), the positive control (lane 7), and the negative control (lane 8).
“Our restoration method is based on the fact that DNA degradation results from random single strand breaks and polymerase reaction restores the nicks, using the other DNA strand as a template”
To obtain a longer restored DNA molecule, we carried out a simple ligation step after the pre-PCR treatment, but the results obtained were no better, using either the TTR1 or the TTR4 primer. Therefore, a ligation step after the pre-PCR restoration treatment is not sufficient to enhance the quality of DNA in postmortem paraffin wax embedded tissue.
Our restoration method is based on the fact that DNA degradation results from random single strand breaks and PCR restores the nicks, using the other DNA strand as a template. The method proposed can be used to obtain longer amplification fragments of around 300 bp of DNA from normally extracted postmortem paraffin wax embedded tissues.
Acknowledgments
The authors are grateful to Dr G Faulkner for the English revision of the manuscript. This work was supported by the contract PSC (263MA108600) from the NIA (NIH).