Article Text

Abstract
Aim: To determine the frequency of tumour budding and somatic APC mutation in a series of colorectal cancers stratified according to DNA microsatellite instability (MSI) status.
Material/Methods: Ninety five colorectal cancers were genotyped for APC mutation in the mutation cluster region (exon 15) and scored for the presence of tumour budding at the invasive margin in haematoxylin and eosin stained sections. A subset was immunostained for β catenin and p16.
Results: The frequency of both somatic APC mutation and tumour budding increased pari passu in cancers stratified as sporadic MSI high (MSI-H), hereditary non-polyposis colorectal cancer (HNPCC), MSI low (MSI-L), and microsatellite stable (MSS). Both budding and APC mutation were significantly less frequent in sporadic MSI-H cancers than in MSI-L or MSS cancers. Tumour buds were characterised by increased immunostaining for both β catenin and p16.
Conclusion: Tumour budding is associated with an adverse prognosis. The lack of budding in MSI-H colorectal cancer may account for the improved prognosis of this subset and may be explained by an intact WNT signalling pathway and/or inactivated p16INK4a.
- colon
- rectum
- cancer
- budding
- APC gene
- mutation
- microsatellite instability
- cdk, cyclin dependent kinase
- DHPLC, denaturing high pressure liquid chromatography
- HNPCC, hereditary non-polyposis colorectal cancer
- MCR, mutation cluster region, MSI, microsatellite instability
- MSI-H, MSI high
- MSI-L, MSI low
- MSS, microsatellite stable
- PCR, polymerase chain reaction
Statistics from Altmetric.com
- cdk, cyclin dependent kinase
- DHPLC, denaturing high pressure liquid chromatography
- HNPCC, hereditary non-polyposis colorectal cancer
- MCR, mutation cluster region, MSI, microsatellite instability
- MSI-H, MSI high
- MSI-L, MSI low
- MSS, microsatellite stable
- PCR, polymerase chain reaction
Prognosis in colorectal cancer has been associated with two morphological features seen at the invasive front of the primary tumour, namely: (1) the low magnification observation of diffuse infiltration and dissection of normal tissues by the cancer1,2 and (2) the high magnification observation of tumour “budding”.3,4 These variables are correlated, with 80% of rectal cancers showing neither feature (64%) or both features (16%).5 The five year survival rate associated with the presence of neither feature was 87%, both features 28%, and one or other feature around 50%.5 Whereas the assessment of diffuse infiltration is subjective and shows considerable interobserver variation, tumour budding is a relatively reproducible feature.5 In a multivariate analysis, tumour budding but not diffuse infiltration was identified as an independent prognostic factor.5
Tumour budding is defined as the presence of isolated single cells or small cell clusters (up to five) that are scattered in the stroma at the invasive margin of the tumour.5 The buds appear to “drip” down from the main mass of more differentiated tumour. The loss of both glandular differentiation and cell cohesion that gives rise to these dissociated elements is probably a crucial event in the development of high invasive and metastatic properties.6 This would be consistent with the important and independent adverse prognostic significance of tumour budding.
“The overexpression of β catenin found in colorectal cancers is secondary to truncating mutation of the APC tumour suppressor gene”
β Catenin has been implicated in the development of tumour budding.7 Accumulation of nuclear β catenin was demonstrated in dedifferentiated, mesenchyme-like tumour cells at the invasive front of colorectal cancers. In the remainder of the tumour, as in normal colorectal epithelium, β catenin is expressed along the lateral cell membrane.8 Analogies between patterning observed at the invasive tumour margin and in embryonic gastrulation have been drawn.8 The overexpression of β catenin found in colorectal cancers is secondary to truncating mutation of the APC tumour suppressor gene. As the principal effector of the WNT signalling pathway, nuclear β catenin is able to complex with the T cell factor family of DNA binding proteins and function as a transcriptional activator.9 Among the target genes activated by β catenin are several known to be crucial in the process of invasion and metastasis. These include urokinase-like plasminogen activating receptor,10 matrilysin,11,12 CD44,13 and laminin 5γ2.14
Based on the model provided by the autosomal dominant condition, familial adenomatous polyposis, it has long been assumed that APC mutation is the initiating alteration in most colorectal cancers.15 In fact, APC mutation is uncommon in small sporadic adenomas,16,17 and the overall frequency of APC mutation in colorectal cancer is around 60%.18 APC mutation is more frequent in rectal than colonic cancer.19 Alternative mechanisms have been suggested for activating the WNT signalling pathway, including oncogenic mutation of the β catenin gene.20 However, this last mechanism is largely restricted to hereditary non-polyposis colorectal cancer (HNPCC).21,22 In most sporadic cancers with a high degree of microsatellite instability (MSI-H) and in a proportion of non-MSI-H cancers, the immunohistochemical expression of tumour β catenin is the same as in normal mucosa, suggesting minimal or no disruption of the WNT signalling pathway.23,24 In keeping with this observation, reliable studies have shown very low frequencies of APC and β catenin mutation in sporadic MSI-H cancer.21,25
It is hypothesised that APC mutation is neither the inevitable initiating event nor an inevitable later event in a subset of colorectal cancers, regardless of MSI status. It is further hypothesised that tumour budding is rare or absent in the subset lacking APC mutation and that APC mutation serves as an important marker of tumour aggressiveness and prognosis. To test this hypothesis, 95 colorectal tumours of known MSI status have been evaluated for both APC mutation and the presence of tumour budding.
MATERIALS AND METHODS
Materials
Most of the tissue samples overlapped with a series of cancers stratified previously on the basis of DNA MSI and investigated with respect to methylation of the DNA repair gene O-6-methylguanine DNA methyltransferase (MGMT). The material included 21 sporadic MSI-H cancers, 43 MSI low (MSI-L) cancers, and 16 microsatellite stable (MSS) cancers. All sporadic MSI-H cancers showed instability in at least one mononucleotide marker (BAT25 or BAT26), instability in at least two of five National Cancer Institute markers,26 and loss of expression of the DNA repair gene hMLH1. The large number of MSI-L cases, in which methylation of MGMT is frequent, is explained by an interest in determining whether there might be an association between MGMT methylation and G : C to A : T transition mutations in APC, as has been described for K-ras and TP53.27,28 Fifteen freshly collected samples of colorectal cancer from patients with HNPCC were also included. Patients were considered to have HNPCC when the Amsterdam criteria were met and at least one cancer in each family was MSI-H with loss of immunohistochemical expression of either hMLH1 or hMSH2. The study was approved by the Royal Brisbane Hospital ethics committee and all participating subjects gave fully informed consent.
Tumour budding
Budding was diagnosed as present or absent on the basis of the findings at the invasive margin in haematoxylin and eosin stained sections. Buds were considered to be present when discrete cell clusters of up to five cells were observed and where this finding appeared to be unrelated to glandular disruption associated with inflammatory cell infiltration. At least five such buds had to be present in a high power field (×40 magnification) (fig 1).

Tumour budding demonstrated by small cell clusters at the invasive margin of a colorectal cancer (haematoxylin and eosin; original magnification, ×40).
Immunohistochemistry
Among the cases were 26 that had been immunostained previously for β catenin (goat polyclonal antiserum; catalogue number sc1496; diluted 1/750; Santa-Cruz Biotechnology, Santa Cruz, San Diego, USA) and scored by a protocol that was not specifically designed to investigate the issue of tumour budding.23 The scoring protocol awarded scores of 1 for loss of cell membrane staining, 1 or 2 for slight and pronounced increase in cytoplasmic staining, respectively, and 1 or 2 for slight and pronounced nuclear staining, respectively, giving a maximum total score of 5. Cases scoring 4 or more were regarded as positive for abnormal β catenin immunolocalisation. Twenty four cases were immunostained for p16 (catalogue number 550834; clone G175–405; dilution, 1/500; BD PharMingen, San Diego, California, USA) with subsequent incubation with peroxidase labelled polymer conjugated goat antimouse immunoglobulin (Envision + System Peroxidase (mouse); Dako, Carpinteria, California, USA), and with colour development using DAB+ chromogen (Dako). Scoring of p16 again was not specifically directed to the issue of tumour budding, with scores of 1 to 4 indicating absent staining, focal staining, patchy staining, and diffuse staining, respectively. Cases scoring 3 or more were regarded as positive.
APC mutations
DNA was extracted from tumour tissue using a salting out technique and amplified by the polymerase chain reaction (PCR) in two separate segments using primers for the “mutation cluster region” (MCR) in exon 15 of the APC gene. To gain maximum specificity a touchdown PCR cycle using the “hotstart” protocol and AmpliTaq Gold (Roche, Applied Biosystems, Foster City, California, USA) was used. Amplified products were subjected to denaturing high pressure liquid chromatography (DHPLC) using optimal running conditions established according to the theoretical melting temperatures calculated on the basis of guidelines provided by Varian (Walnut Creek, California, USA; at http://insertion.stanford.edu/melt.html) and also on the basis of the melting profiles calculated manually on the instrument for each PCR fragment. The high sensitivity of the DHPLC technique was validated previously by testing samples with known mutations. After the identification of aberrant peaks, manual sequencing was undertaken to determine an underlying mutation. Mutations were scored as positive only when manual sequencing indicated the presence of substitutions, deletions, or insertions that generated stop codons. Cancers with more than one APC alteration were scored simply as mutation positive for the purposes of our study.
Statistical analysis
The data were analysed by χ2 analysis and Fisher’s exact test.
RESULTS
DNA samples from 14 of 21 MSI-H cancers showed one or more aberrant peaks with DHPLC. Subsequent manual sequencing disclosed 12 cases with at least one non-functional change in the APC gene, although only one aberrant peak was associated with a truncating mutation (table 1). Thirteen of 15 HNPCC cancers had one or more aberrant peaks with DHPLC. Four of these had at least one non-functional alteration, three showed a single truncating mutation, and a further specimen had two truncating mutations in APC (table 1). Of 43 MSI-L cancers, 28 had at least one aberrant peak with DHPLC. Of these, eight had at least one non-functional alteration and 14 had one truncating mutation in APC (table 1). Thirteen of 16 MSS cancers had at least one aberrant peak with DHPLC. In these, at least one non-functional alteration was found in four samples, whereas a single truncating mutation was detected in six samples (table 1). Most of the non-functional alterations were A to G transitions at codon 1494 resulting in no amino acid change (a frequent germline polymorphism). Overall, 56% of the APC alterations were regarded as non-functional and these were distributed with a similar frequency in samples stratified by MSI status. Seven of the total of 14 stop mutations in MSI-L cancers resulted from deletions, mainly in short polyadenine sequences. G : C to A : T transitions occurred in only one MSI-L cancer.
Spectrum of somatic APC mutations according to DNA microsatellite status
The frequency of somatic APC mutation was significantly lower in sporadic MSI-H cancers (4.8%) than in MSI-L (32.6%; p = 0.01) and MSS cancers (37.5%; p = 0.01) cancers. The frequencies of tumour budding and of APC mutation were similar in terms of their patterns of distribution across the tumour subgroups (table 2). The frequency of tumour budding in sporadic MSI-H cancers (0%) was significantly lower than the frequency of budding in MSI-L (41.9%; p < 0.001) and MSS (56.2%; p < 0.001) cancers. Budding was also less frequent in HNPCC (20%) than in MSS cancers (p = 0.04; table 2).
Frequency of APC mutation and budding in colorectal cancers stratified as MSI-H, HNPCC, MSI-L, and MSS
Even though the scoring of β catenin and p16 immunostaining was not specifically directed at tumour buds, positive findings were more common in cases with budding. In the case of β catenin, positive staining in buds was both nuclear and cytoplasmic (fig 2). Positive staining was recorded in six of nine cases with budding, but only four of 17 cases without budding, the difference being significant (p = 0.03). Six of the 26 cases stained for β catenin had a pathogenic APC mutation. Of these, five scored as positive and the sixth had the highest possible negative score (3). No APC mutations were detected in cancers with a negative score for β catenin. Five cancers with a positive β catenin score were negative for APC mutation. With respect to p16 immunohistochemistry, staining within buds was both nuclear and cytoplasmic but mainly cytoplasmic (fig 3). Positive staining for p16 was recorded in six of 11 cases with budding, but only three of 13 cases without budding, with the difference falling short of significance (p = 0.11).

Extensive cytoplasmic and nuclear immunostaining for β catenin in tumour buds (avidin–biotin complex technique).

Tumour buds showing cytoplasmic expression of p16 (avidin–biotin complex technique).
DISCUSSION
The overall low frequency of cancers with one or more APC mutation (22%) is explained in part by the over representation of sporadic MSI-H and HNPCC cases in this series and in part by limiting the search for APC mutation to the hot spot MCR in exon 15. It could be argued that the low frequency of APC mutation (4.8%) in sporadic MSI-H results from the fact that APC mutation occurs outside the MCR in this subset. This interpretation is unlikely for several reasons. The observation fits with more extensive studies of APC mutation in colorectal cancers, in which sporadic MSI-H cancer was carefully distinguished from HNPCC.21,25 The finding also fits with the frequent retention of a normal β catenin immunolocalisation pattern in this subset.23,24 The frequency of APC mutation in HNPCC cancer is also low (26.7%). However, within this subgroup, disruption of the WNT signalling pathway may also occur through mutation and oncogenic activation of β catenin.22 Aberrant immunolocalisation of β catenin within HNPCC cancers29 fits with the mutational evidence for WNT pathway disruption.
“These findings indicate that budding is a dynamic process under genetic control and not merely the result of architectural disruption caused by a host immune reaction at the tumour margin”
The frequency of APC mutation in the MSI-L group (32.6%) differed from the MSI-H group (4.8%), but not from MSS (37.5%) cancers. Although the difference between MSI-L and MSS cancers was non-significant, the trend across the cancer subgroups fits with findings for other markers of WNT pathway disruption—specifically, loss of heterozygosity at 5q and aberrant immunolocalisation of β catenin.23 Contamination of the MSI-L group by MSI-H cases is an unlikely explanation for the low frequency of APC mutation in the MSI-L group or the intermediate findings (between MSI-H and MSS cancers) with respect to tumour budding. None of the MSI-L cancers in our study showed either instability at mononucleotide markers or loss of expression of DNA mismatch repair proteins (data not shown). Furthermore, we have shown previously that MSI-L cancers are distinguished from MSI-H cancers by a high frequency of methylation of the DNA repair gene MGMT and a high frequency of K-ras mutation.30 Methylation of MGMT has been associated with G : C to A : T transitions in K-ras and TP53.27,28 Although most of the MSI-L cancers in our study showed methylation of MGMT,30 50% of APC mutations were deletions on short polyadenine tracts and only one of 14 truncating mutations was a G : C to A : T transition. This suggests that in a subset of MSI-L neoplasms, methylation of MGMT may be acquired in adenomas subsequent to early APC mutation. Large selection pressures must account for the frequent finding of frameshift APC mutations in tumours that are not otherwise prone to instability in mononucleotide tracts.
The frequencies of tumour budding and of APC mutation were distributed in similar patterns across the four subsets of colorectal cancer (table 2). Within the subsets, however, there was no correlation between budding and APC mutation. In the case of MSI-H and HNPCC cancers, both features were uncommon. Within the MSI-L and MSS groups were cancers with budding but no APC mutation, and vice versa. Combining these groups, budding occurred in 20 of 39 cases without mutation and seven of 20 cases with mutation. However, it is inevitable that the frequency of APC mutation is underestimated in both groups. In addition, the frequency of budding in both groups was high compared with other studies and may have been overestimated. For example, the frequency of budding in a series of rectal cancers (which would have been unlikely to have included many sporadic MSI-H or HNPCC cancers) was around 30%.5 A further explanation for the discordant findings may be the prerequisite for factors other than a single APC mutation (for example loss of heterozygosity at 5q) or a fully independent mechanism (see below). Budding was present in the single cancer (an HNPCC cancer) with two (presumably biallelic) truncating mutations.
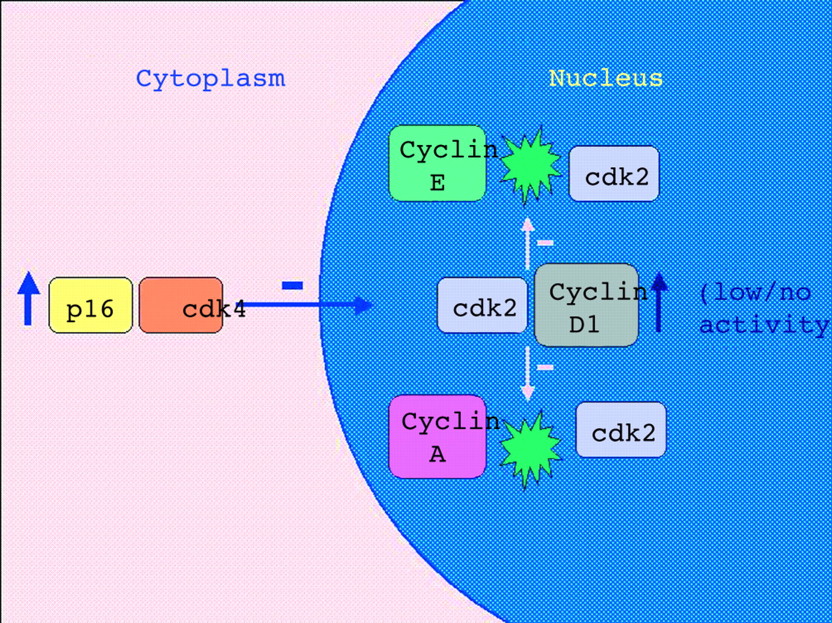
In our study, a subset of cancers was immunostained for β catenin and p16. As has been shown by others, the invasive margin and specifically tumour buds showed increased expression of β catenin and p16.7,8 The increased expression of β catenin was both cytoplasmic and nuclear, whereas p16 expression was mainly cytoplasmic. These findings indicate that budding is a dynamic process under genetic control and not merely the result of architectural disruption caused by a host immune reaction at the tumour margin. Tumour budding is known to be associated with reduced proliferation, yet nuclear cyclin D1 expression is increased.8 The increased cytoplasmic p16 expression may explain this paradox. For example, p16 may bind cytoplasmic cyclin dependent kinase 4 (cdk4) and block its nuclear translocation. Cyclin D1 is known to form relatively inert complexes with cdk2 and would therefore, in the absence of nuclear cdk4, compete with cyclins A and E (fig 4).31 Although the preceding model is speculative and depends upon the demonstration of cytoplasmic binding of cdk4 by p16, it provides an economical explanation for the discordant findings of raised p16, raised cyclin D1, and lowered proliferation at the invasive margin of colorectal cancer.8 It is possible that the expression of p16INK4a and WNT pathway disruption may serve as joint prerequisites for tumour budding. The lack of tumour budding in MSI-H cancers would be explained by the known silencing of p16INK4a through methylation of the promoter region of the gene,32 or by the lack of WNT pathway disruption (or both). There was no correlation between the methylation of p16INK4a and budding among the non-MSI-H cancers in our study (data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dysregulated p16 binds cyclin dependent kinase 4 (cdk4), which results in cytoplasmic sequestration of cdk4. Binding between nuclear cyclin D1 and cdk2 results in an inert complex but serves to compete with the binding of cdk2 to cyclins A and E. This may explain why there is increased expression of nuclear cyclin D1 in tumour buds whereas proliferation is decreased.
In summary, the feature of tumour budding has been associated with invasion, metastasis, and poor prognosis. The findings of our present study suggest that the lack of this feature in sporadic MSI-H colorectal cancer, in turn explained by an intact WNT signalling pathway and/or methylation of p16INK4a , may serve as the explanation for the low biological aggressiveness of this subgroup.
Take home messages
-
Tumour budding is associated with invasion, metastasis, and an adverse prognosis
-
The lack of budding in microsatellite instability high colorectal cancer may account for the improved prognosis of this subset and may be explained by an intact WNT signalling pathway and/or inactivated p16INK4a
Acknowledgments
The study was supported by the NCI Colon Cancer Family Registry, NCI/NIH, grant no. 1-U01-CA74778.