Article Text

Statistics from Altmetric.com
- QTc, heart rate corrected QT interval
- SIDS, sudden infant death syndrome
- SUNDS, sudden unexpected nocturnal death syndrome
A discussion of the evidence to date
Long QT syndrome causes sudden unexpected death through rapid ventricular tachycardia (“torsades de pointes”) leading to ventricular fibrillation (see figs 1 and 2).1,2 The postmortem examination reveals no cause for the death. These characteristics make long QT syndrome a plausible cause of sudden infant death syndrome (SIDS), and many research efforts have been made to investigate a possible link. The genetic forms of long QT syndrome are usually inherited in an autosomal dominant fashion (Romano-Ward syndrome). Yet gene carriers can be asymptomatic, and around a third can even have a normal ECG.3 Therefore the condition might theoretically cause multiple SIDS within a family, with apparently healthy parents. It has been suggested that parents in such families might have been wrongfully accused of murder.4,5

(A) This six lead rhythm strip from an infant with syndactyly and long QT syndrome shows gross prolongation of the QT interval. Two p waves are seen for each T wave. There is 2:1 atrioventricular block because the t wave has not even started when the next p wave appears; since ventricular repolarisation has not yet occurred, the ventricle cannot yet be depolarised again. This phenomenon has been reported in sudden infant death secondary to long-QT type 3.37 (B) This six lead rhythm strip was taken from the same infant as in (A), moments later. The QT interval is very long, and the t wave is alternately upright and then inverted. Repolarisation is disordered and swinging like a pendulum in one direction across the myocardium and then another. This typically precedes torsades de pointes.

{kind=link}
{kind=link}
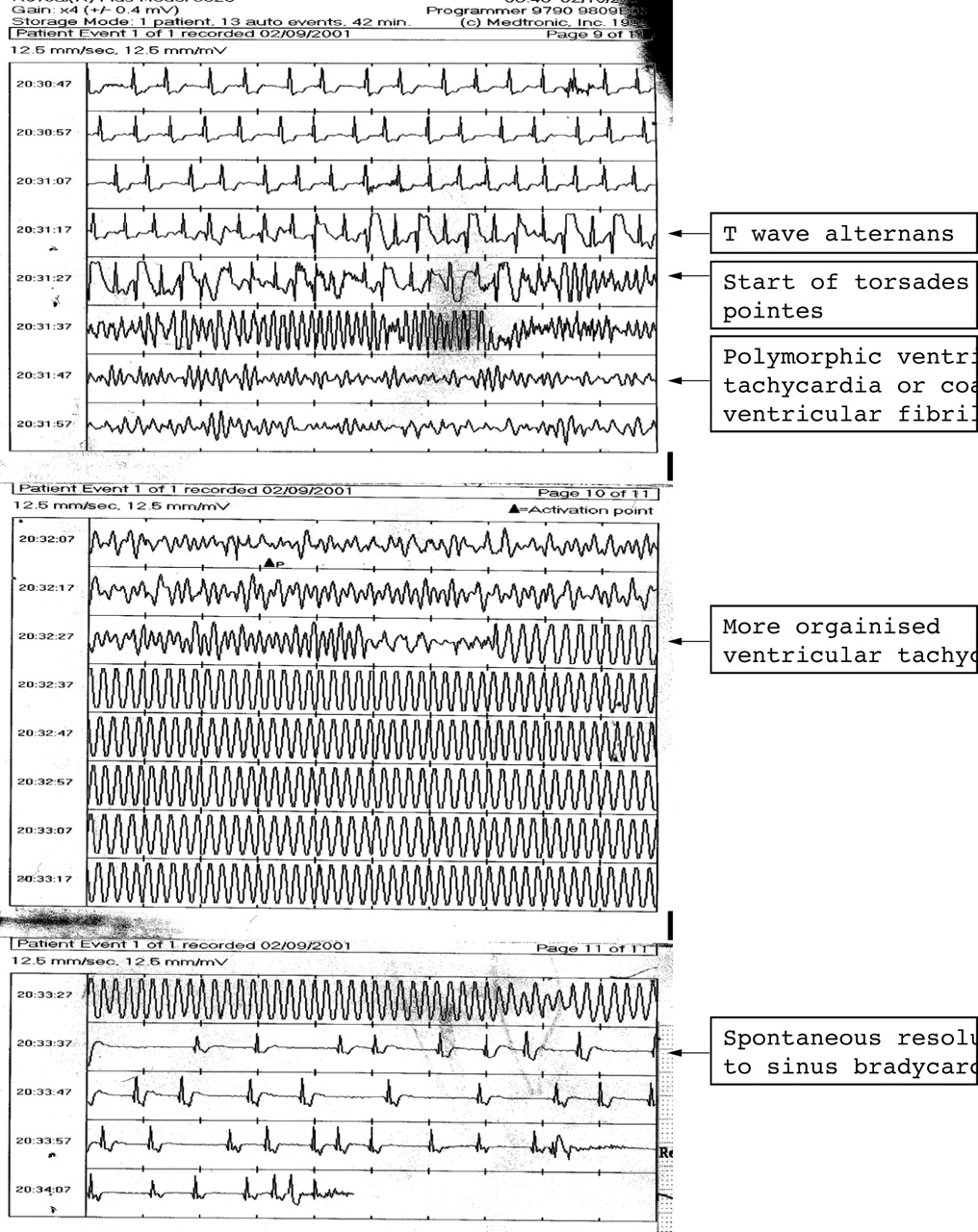
ECG strip recorded from an implanted digital loop recorder (Reveal device) during a syncopal episode from which recovery occurred spontaneously. Although taken from an 11 year old boy, this shows features which are instructive and relevant to an infant with long QT syndrome. T wave alternans precedes torsades de pointes. The torsades initiates when a ventricular depolarisation (QRS complex) occurs during the preceding T wave. The torsades can occur because a repolarisation gradient has developed between different parts of the myocardium. A rapid re-entry circuit is established, causing rapid ventricular tachycardia which moves around the heart so that the morphology of the tachycardia changes in a cyclic fashion. The ventricular rate is so rapid that the patient loses consciousness immediately. As indicated by the arrows, the tachycardia becomes more organised before reverting spontaneously to sinus rhythm, 30 seconds after the tachycardia started.
PROLONGATION OF THE QT INTERVAL, TORSADES DE POINTES, AND SUDDEN DEATH
If ventricular repolarisation (represented by the QT interval on the ECG) is prolonged or disordered, there is a risk of torsades de pointes leading to ventricular fibrillation when a ventricular extra beat occurs during repolarisation. QT prolongation can occur due to a genetic abnormality or secondary (acquired) to a variety of factors including medications (such as cisapride or terfenadine), myocardial disease, biochemical factors (particularly hypokalaemia), or neurohumoral and physiological influences (such as hypothermia and severe brain injury).
The inherited forms of long QT syndrome are mostly secondary to a dysfunction in the transport of potassium and sodium ions through channels across the myocardial cell membrane. This leads to prolongation of the myocardial action potential, finally expressed as prolongation of the QT interval on the surface ECG.6 Acquired QT prolongation (sometimes called acquired long QT syndrome7,8) is also the result of these same channels becoming dysfunctional.
GENETIC FACTORS (INHERITED LONG QT SYNDROME)
Seven genotypes of long QT syndrome have been identified so far.2 The genes encode complex proteins forming the ion channels or assisting in their function. They account for about two thirds of families with long QT syndrome; one third of long QT families have yet to have a genetic mutation identified.9 Table 1 shows the features of the three commonest types (accounting for 95% of the known mutations). Types 1 and 2 are the most common forms, of similar prevalence, and are the result of underactive potassium channels. Type 3 accounts for about 7% of genotyped families, and is the result of an overactive sodium channel (INa, encoded for by the gene SCN5A). Two to three per cent of genotyped families carry two mutations. Types 4 to 7 are rare, though the true incidence of type 4 is uncertain since the mutation (Ankyrin B) has only recently been discovered. The condition of syndactyly with long QT syndrome is a very severe form of the disease, caused by a calcium channel dysfunction.10 Jervell and Lange-Neilsen syndrome is also very severe, due to the co-inheritance of two potassium channel defects which also results in deafness, and presents with an autosomal recessive type of inheritance.11
Each of the genotypes has its own phenotype expressed as characteristic T wave abnormalities on the ECG and by the type of trigger for syncope or sudden death.12 There is considerable overlap between the phenotypes; however, syncope or sudden death are classically triggered by physical exertion (especially swimming) in long QT type 1, and emotional excitement or loud noise (especially noises causing waking from sleep) in type 2. Risk of sudden death in types 1 and 2 is greatly reduced with regular beta blocker medication. With type 3, death most commonly occurs during quiet rest or sleep, and syncope is relatively uncommon; the first event is often sudden death.12 These characteristics make long QT type 3 a most likely candidate to cause SIDS. Other mutations within the same sodium channel encoding gene (SCN5A) can result in under activity of the sodium channel, and cause Brugada syndrome or SUNDS (sudden unexpected nocturnal death syndrome).13 Death in these syndromes also occurs at night due to ventricular fibrillation, and the QT interval is normal.
NON-GENETIC FACTORS CAUSING PROLONGATION OF THE QT INTERVAL
Medications
The list of medications which can either prolong the QT interval or trigger an episode of torsades is large, and is frequently updated on a website dedicated to this (www.qtdrugs.org). Epidemiological studies of SIDS have shown some link to sedative medications, but not yet to ingestion of QT prolonging medications.
Physiological factors
Quiet sleep prolongs the QT interval.14 In the light of the success of supine sleeping position in reducing SIDS, two studies looked at the effect of prone positioning on QT interval at 1 month of age. One study showed an increase in QT intervals and reduced heart rate variability,15 and the other showed no difference.16 Intriguingly however, one infant died of SIDS two months after the latter study; this infant had shown a rise in QT interval of 25 ms lying prone. The authors postulated that only infants with long QT syndrome might show this effect.
Physical and emotional stress can trigger syncope and death in long QT syndrome. Precipitants of stress in the newborn include pain, hunger, heat (trapped under the duvet with the head facing the mattress), and also episodes of minor upper airways obstruction. Although these might trigger ventricular tachycardia in susceptible individuals, it is striking that ventricular arrhythmias have not been seen in large cohort physiological studies of infants with upper airway obstruction.17
The peak of SIDS at 2–3 months of age coincides with the period when the heart rate corrected QT interval (QTc) tends to be longest after the first week of life.18,19 It has been postulated that this QT prolongation represents a transient developmental imbalance between the innervation by the right and left sympathetic nerves such as has been postulated to be factorial in long QT syndrome.20,21 However, this hypothesis is weakened by the observation that the QT interval is the same in the first few days of life when SIDS is very uncommon.
Central nervous system abnormality
Cerebral injury at all ages can cause QT prolongation.22 The brain stem of SIDS victims sometimes show abnormalities of medullary nuclei, which are involved in both respiratory and circulatory control.23 This, or other central nervous system abnormalities could conceivably affect the QT interval.
STUDIES INVESTIGATING A LINK BETWEEN SIDS AND LONG QT INTERVALS
In 1976 Maron et al studied ECGs on 42 sets of parents who had an infant with SIDS, and found that 11 (26%) of the sets had at least one with a prolonged QT interval.24 Since then there has been a small number of infants reported in the literature who died suddenly, but serendipitously had an ECG beforehand which showed a long QT interval or even ventricular tachycardia. For example, in 1979 Southall et al described an infant who died at 12 days, with a preceding ECG and a very long QTc of 630 ms, and with 2:1 atrioventricular block secondary to the prolonged ventricular repolarisation.25 In 1980 Di Segni et al described an infant with torsades de pointes on day 1.26
PROSPECTIVE EVALUATION OF ECGS IN THE NEWBORN
The largest prospective study of neonatal ECGs was published by Schwartz et al in 1998.27 The data were collected from nine maternity units in Italy between 1976 and 1994, including 34 442 infants on day 3 or 4 of life. Twenty four subsequently died of SIDS (a remarkably low incidence for this time period; 0.7/1000). Mean QTc of a random sample of the non-SIDS (9725 ECGs) was 393 ms with 97.5 centile being 440 ms. The mean QTc of SIDS infants was 435 ms, with 12 (50%) above 440 ms. Three had values greater than 470 ms. The study was criticised methodologically.28 In addition, the low incidence of SIDS deaths seen in this study at that time leads to the suspicion that there may have been a bias in ascertainment. This is the only population based evidence to date that suggests long QT interval is a risk factor for SIDS in a significant proportion of infants. Previous, smaller studies by Southall et al and Weinstein et al found no difference between SIDS and controls.29–31 Taking together the 47 ECGs from infants who subsequently died of SIDS from these combined studies, 15 of 47 (32%) had a QTc over 440 ms.
MOLECULAR GENETIC EVIDENCE IN SIDS VICTIMS
Ackerman et al screened for long QT gene mutations in DNA stored from 93 SIDS cases at postmortem examination.32,33 Four of the 93 cases had pathological mutations (4.3%; 95% CI 1.2% to 10.7%), two within SCN5A and two within each of the potassium channel genes associated with long QT 1 and 2 respectively. Thus it is likely that these four infants died due to long QT syndrome. The true figure may be higher since more than a third of long QT syndrome families have yet to have the abnormal gene identified, and so cannot be screened for in this manner. Therefore if this sample proves to be representative, between 6% and 7% of SIDS may be due to the genetic form of long QT syndrome. The incidence of SCN5A mutations in the general population is not known. However, the same research group report a high incidence of potassium channel mutations among 744 apparently healthy adults (25% in black and 14% in white Americans).34 These polymorphisms occurred in 25% of the SIDS group too. Cellular electrophysiological testing has recently shown that some of these polymorphisms in fact lead to some mild channel dysfunction in the laboratory, and may not therefore be harmless variations in genetic structure.35 Such minimal dysfunction with common polymorphisms can be expressed clinically with torsades de pointes when QT prolonging medication is given.7 Whether extreme physiological stress can do the same in the infant is not yet known.
Identifying mutations in SIDS victims does not necessarily prove causality, even if they are known to be pathogenic in other families, or on cellular testing. The infants identified by Ackerman et al still may have died of causes other than rapid ventricular tachycardia or ventricular fibrillation. However, three recent case reports have shown such mutations, all in SCN5A, in infants with documented ventricular tachycardia or ventricular fibrillation. Wedekind et al reported an infant who died at 9 weeks with a previously documented polymorphic ventricular tachycardia and a 12 lead ECG with a QTc of 600 ms.36 Schwartz et al reported a 44 day old infant who had a sudden cardiac arrest with ventricular fibrillation and torsades de pointes, who was successfully cardioverted.37 The QTc was 648 ms. Both of these cases occurred de novo; the mutation was not found in either parent. In this issue, our group in New Zealand report an infant who, having previously been asymptomatic, had a cardiac arrest with documented ventricular fibrillation at 19 days. Remarkably, the resting ECG is normal, with a normal QT interval. He and his asymptomatic mother have a mutation within SCN5A previously seen in a family with sudden unexpected nocturnal death syndrome.38 In addition to these cases, Schwartz et al recently reported a SIDS case in whom they identified a mutation in KVLQT1 (long QT type 1).39
Irrefutable evidence therefore exists that SIDS is due, in a minority, to cardiac channelopathies such as long QT syndrome.
If long QT syndrome is not an uncommon cause of SIDS, why have so few episodes of ventricular tachycardia been documented by those investigating SIDS, and why have QT measurements not been long in near miss SIDS?40
The answer may be that non-fatal (“syncopal”) events are rare, such as with long QT type 3. Perhaps infants investigated for “near miss” sudden death episodes are less likely to have long QT type 3 than other conditions, since they would have died with the first cardiac event. Nevertheless, the few infants who have had heart rate monitors attached at the time of death at home have to date shown bradycardia, and not ventricular tachycardia.41 Furthermore, it is important to retain the perspective of the huge body of epidemiological and pathological data which tends to suggest respiratory and particularly upper airway pathology as a primary cause of SIDS.42–46
SHOULD THE ECG BE USED TO SCREEN FOR LONG QT SYNDROME IN INFANTS?
Analysis of the data from the mass neonatal ECG screening study of Schwartz et al shows that a QTc of above 440 ms would have a positive predictive accuracy for SIDS of only 1.4%.28 Furthermore, accurate measurement of neonatal QT interval is difficult, with 95% confidence limits for repeatability as high as 50 ms in infants.30 At such rapid heart rates, determination of the end of the T wave can be difficult, and correction for heart rate using the Bazett formula (QTc = QT interval divided by the square root of the preceding R-R interval) is invalid.47 Positive and negative predictive accuracy would therefore be even worse than 1.4%.
Schwartz et al estimated, from their mass neonatal ECG screening study, that 100 infants would have to be treated (with β blockers) to save two lives.27 However, even if the ECG is positive, therapy with β blockers may be ineffective in reducing risk. Long QT type 3, the commonest type in SIDS to date, is not proven to respond to β blockers.48 It would be counter-intuitive if they did, since events occur at rest, and not at times of adrenaline release—unlike types 1 and 2.
The question is not only whether the “economic inefficiencies of current screening methodologies supersede the value of a young life”,49 but also whether such an ECG based screening programme would affect outcome at all.
MULTIPLE SIDS WITHIN A FAMILY
Multiple SIDS within a family may theoretically be caused by long QT syndrome, but there is no confirmatory evidence to date. Some (though not all) of the SCN5A mutations cause severe, sporadic forms of long QT syndrome which are not present in other family members.36,37 Furthermore, it is the norm among families with long QT syndrome to have a profoundly variable clinical picture with the same genetic mutation; one member may have a cardiac arrest and gross QT prolongation, and another have no symptoms and a normal ECG. Hitherto unknown factors vary clinical expression within a family. It would therefore seem to be improbable, though not an impossible rare event, that long QT syndrome would cause three sibs to die in infancy within one small family. A postmortem molecular study of long QT genes in such cases should help clarify this, as well as careful evaluation of ECGs in their extended families.
POSTMORTEM INVESTIGATION OF SIDS
A priority of the investigation of postmortem negative sudden death in the young adult is to discover cardiac inherited diseases which might cause the death of other family members. These are found in about a quarter of families.50 This is achieved by taking a careful family history and analysing the 12 lead ECGs of all first degree relatives. Although the original study of ECGs of parents of SIDS victims by Maron and colleagues24 suggests a similar proportion would be found following SIDS, current molecular evidence suggests this will be much lower. Nevertheless, assuming familial arrhythmic syndromes are confirmed to cause a small but significant proportion of SIDS, the postmortem investigation will provide an important opportunity to detect such families, and instigate therapies which can save very many lives in their extended families. Molecular diagnosis is not uniformly available; furthermore it cannot be relied on alone since more than a third with definite long QT syndrome have no DNA diagnosis available yet. For the time being, therefore, it would seem prudent (and inexpensive), to ascertain a careful family history and ECGs from first degree relatives of SIDS victims. Cooperation is essential between coroners, forensic pathologists, paediatricians, and cardiologists to achieve such screening effectively. Practitioners interviewing the family need to be suspicious of exercise or emotionally induced syncope or sudden death, unexpected drownings, “familial epilepsy”, nocturnal seizures, and possibly multiple SIDS within the family.
DNA should be stored from the body at postmortem examination, and the family should be informed of this by the coroner. If tissue is not retained, the neonatal screening (Guthrie) card can be a useful source of DNA.51 If all infectious, metabolic, and other tests are negative, it is ethical and reasonable to seek a molecular diagnosis of long QT syndrome.
This approach will not greatly reduce the incidence of SIDS, since the vast majority are not due to an arrhythmic syndrome. However, in some it will give families an explanation for the loss of their baby, and physicians and coroners an opportunity to prevent more sudden deaths at all ages within families who have as yet undetected inherited arrhythmic syndromes.
CONCLUSIONS
Molecular evidence suggests long QT syndrome, or other sudden cardiac death syndromes, may cause about 5% of SIDS.32,33 ECG based screening of parents of SIDS victims24 suggests this incidence may be higher. More studies using both techniques are needed. Mutations within SCN5A, causing long QT type 3 and the sudden unexpected nocturnal death syndrome, make this the most likely candidate gene. However, mutations causing long QT types 1 and 2 are more common in the general population and may prove to be equally important in SIDS with further study of larger and more divergent populations.
Future research may identify a link to milder forms of long QT syndrome, analogous to the heterogeneic response of the population to drugs known to prolong the QT interval.7 However, infants usually respond to physiological stress by extreme bradycardia rather than ventricular tachycardia.
The ECG is a remarkably poor test to screen for SIDS, and the most frequently recognised long QT genotype in SIDS has not been shown to respond beneficially to β blockers. The ECG should not yet be used for mass neonatal screening to prevent SIDS, since there is insufficient evidence that it will.
Acknowledgments
For their helpful comments I am most grateful to Drs Ed Hey, Karen Gray, John French, and Professor Mark Rees.
A discussion of the evidence to date
REFERENCES
Footnotes
-
Competing interests: none declared