Article Text

Statistics from Altmetric.com
- APC, adenomatous polyposis coli
- BMDC, bone marrow-derived stem cell
- COX2, cyclooxygenase 2
- GORD, gastro-oesophageal reflux disease
- LOH, loss of heterozygosity
- MAPK, mitogen-activated protein kinase
- PPI, proton pump inhibitor
- TGFβ, tumour growth factor β
Barrett’s oesophagus is the eponym used to describe the change from the normal stratified, squamous epithelium of the lower oesophagus to a polarised, columnar-lined epithelium with intestinal-type differentiation.1 This condition develops in the context of chronic gastro-oesophageal reflux disease (GORD)2 and is associated with a 0.5–1% annual conversion rate to oesophageal adenocarcinoma.3,4 The overall 5-year survival rate in patients presenting with symptomatic adenocarcinoma is a dismal 13%.5 An understanding of the molecular basis for the development and progression of Barrett’s metaplasia is required to develop effective clinical management strategies. Before discussing the molecular changes at the level of the tissue, it is important to briefly consider the environmental and inherited genetic factors that contribute to an individual’s susceptibility to these conditions.
EFFECT OF ENVIRONMENTAL AND INHERITED FACTORS ON INDIVIDUAL SUSCEPTIBILITY
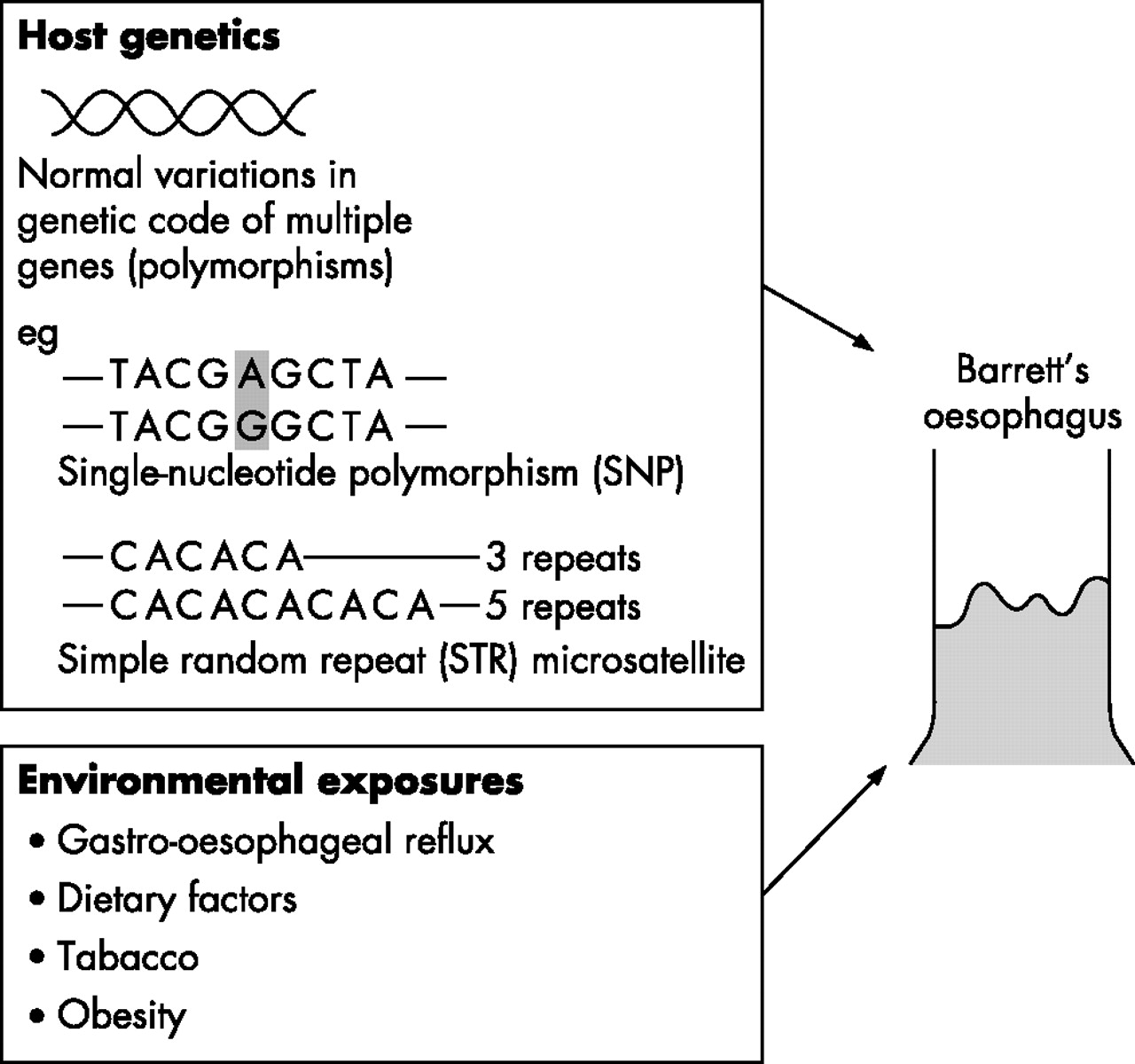
The role of environmental factors is evident from the short time period over which the incidence of Barrett’s oesophagus6 and oesophageal adenocarcinoma7 has increased. Furthermore, the demonstration of a “birth cohort effect”, with higher incidence rates in younger cohorts,8 would support the idea that exposure to environmental factors in early life is an important determinant of risk. Identification of specific environmental exposures is difficult, but factors that increase gastro-oesophageal reflux, such as dietary components, increasing body mass index and eradication of Helicobacterpylori, may be relevant.9 Whether or not smoking and alcohol consumption are risk factors for Barrett’s oesophagus is controversial; however, an association was found in a recent population study.10 In order for an individual to develop Barrett’s oesophagus, and in some cases oesophageal adenocarcinoma, these environmental exposures probably need to interact with genetically determined characteristics that define personal susceptibility11 (fig 1). As most cases are sporadic, occurring in the absence of a family history, these inherited genetic factors are likely to be normal variations or polymorphisms in multiple genes rather than single gene mutations. In Barrett’s oesophagus, polymorphisms in genes involved in DNA repair, chemical detoxification and cytokine responses have been identified.12–14 However, to reliably detect the low to moderate risks expected to be associated with multiple genetic polymorphisms, large, population-based case collections are required, with comprehensive epidemiological, clinical and pathological data.15 In summary, Barrett’s oesophagus occurs in the context of inherited genetic susceptibility loci and specific environmental exposures. The remainder of this review is concerned with the molecular changes in the oesophageal tissue in the development and progression of Barrett’s oesophagus.

The likelihood of an individual developing Barrett’s oesophagus might be determined by a combination of host genetics and environmental factors. The host genetics will include normal variations in multiple genes including polymorphisms and microsatellites. Examples of these changes are shown. The specific genes involved and the identification of particular environmental factors are still being elucidated.
INDUCTION OF BARRETT’S METAPLASIA
The process of metaplastic change is rarely observed in vivo and there are no reliable, physiological animal models. As a result, the theories attempting to explain the molecular and cellular processes underlying the development of Barrett’s oesophagus are rather speculative. However, it has been established that the luminal environment might be important. Cells of the human oesophageal epithelium are under relatively unique environmental pressures, being exposed on a daily basis to thermal stress, unmetabolised chemicals or food products. For Barrett’s oesophagus to develop, chronic exposure to refluxed duodenal and gastric juices seems to be critical. Hence, all of the proposed theories for the origin of Barrett’s oesophagus have in common the suggestion that luminal damage of the epithelium is required at the outset. It has also become apparent that Barrett’s metaplasia is likely to originate from within the oesophageal compartment rather than from overgrowth of neighbouring gastric tissue, as animals can still develop columnar oesophagus when there is a mucosal defect separating the distal oesophagus from the transitional zone.16,17
Cell of origin
The metaplastic conversion of the oesophageal squamous epithelium to a columnar-lined epithelium could arise from two different categories of cell. One possibility is the direct conversion of differentiated cells in the absence of cell proliferation, a process called “transdifferentiation”. Alternatively, metaplasia may develop from the conversion of a “stem” or “pluripotential cell”, meaning a cell with the capacity for unlimited or prolonged self-renewal18 (fig 2).

Possible cells of origin for Barrett’s metaplasia. If the metaplastic process occurs by altered differentiation of a mature squamous oesophageal epithelial cell (shaded cells in grey in the mucosal layer) without requiring proliferation, it is called transdifferentiation. Alternatively, the cell of origin may be an undifferentiated cell with the capacity to form multiple-cell lineages: a so-called “stem cell”. These stem cells may be of tissue or bone marrow origin. The tissue-derived stem cells may be located in the basal compartment of the interpapillary layer or in the submucosal gland duct. In either case, the trigger for columnar differentiation seems to depend on surface epithelial damage from luminal factors.
Recent evidence for transdifferentiation has come from in vitro culture of embryonic mouse oesophagus. These experiments take advantage of the normal developmental process whereby the oesophagus undergoes a columnar to squamous cell transition at 18 weeks of gestation.19 The evidence suggests that the cells in the squamous basal layer arise directly from columnar tissue, independent of squamous cell proliferation or apoptosis of columnar cells.20 An extrapolation of this work is that in adulthood the reverse transdifferentiation process could account for the generation of Barrett’s metaplasia in the context of GORD. However, the embryological maturation event may be quite different from the pathological development of metaplasia. Evidence against transdifferentiation comes from the observation that new squamous epithelium can develop after ablation treatment in which the Barrett’s epithelium has been completely eliminated.
With regard to the stem cell theory, the squamous oesophageal stem cells are thought to reside in the interbasal layer of the epithelium between the papillae.21 Possibly, there are stem cells in the glandular neck region of oesophageal submucosal gland ducts similar to those found within the bulge region of the hair follicle.22 Hence, after ulceration or damage, stem cells may grow out to form a new gland in the lamina propria, finally giving rise to a duct by which the glandular cells are carried to the surface.16,23 The basis for this mechanism is the ulcer-associated cell lineage.24 Whether the normal squamous and Barrett’s epithelial cells arise from the same or different progenitors is not clear. However, recent mutational data that examine squamous islands compared with the surrounding Barrett’s epithelium suggest that in most cases the progenitor cells seem to be different for each tissue type.25
As well as tissue-specific stem cells, it is now known that bone marrow-derived stem cells (BMDCs) have such a surprising degree of plasticity that they could also give rise to diverse epithelial cell types.26 Bone marrow-derived epithelial cells have been identified throughout the gastrointestinal tract 11 months after transplantation of a single purified haematopoietic BMDCs.26 The local tissue environment, such as continued inflammation and injury, seems to have an important role in this process.27 This concept is well illustrated by the demonstration of BMDCs in the murine stomach after Helicobacter-induced chronic gastritis. These BMDCs had a classical metaplastic phenotype, which gradually progressed to dysplasia and neoplasia.28 Obvious parallels can be drawn between this model and Barrett’s carcinogenesis, although there is no direct evidence at the current time for the role of BMDCs in this disease. Whether the progenitor cell arises from within tissues or the bone marrow, the stem cell origin would more easily explain the perpetuation of the metaplastic phenotype once it had occurred.
Determinants of cell fate
The control of cell fate is likely to be achieved by a combination of internal and external cell signals. Internal signals may include nuclear factors controlling gene expression. External signals are likely to be reflux components, secreted factors and the factors responsible for cell–cell adhesion, such as extracellular matrix proteins. One possibility is that genes responsible for the physiological transition from columnar to squamous oesophagus in embryogenesis may be abnormally reactivated, leading to the development of metaplastic Barrett’s oesophagus in adulthood. For example, in the embryo, a morphogenic gradient may induce activation of specific genes resulting in one tissue type (eg, gastric epithelium), whereas non-induction of those same genes may lead to a completely different tissue (squamous oesophageal epithelium). If pluripotential cells of the squamous oesophagus still require inactivation of the same genes in adult life to maintain their stratified squamous differentiation structure, then re-activation of those genes may lead to an area of columnar metaplasia29 (fig 3). One example in support of this theory is the conversion of pancreas cells to liver cells, which can be achieved by induction of a single transcription factor.30 It is not yet known whether a similar process occurs in the oesophagus.

The molecular mechanism for Barrett’s metaplasia may result from change in the activation status of a gene as a result of injury. The example shown here is activation of gene “x” in the oesophagus. This hypothetical gene would ordinarily be switched off during embryogenesis when the oesophagus changes from a columnar-lined epithelium to a squamous epithelium. Reactivation of that gene may lead to a patch of columnar tissue, which may then grow to occupy the lower part of the oesophagus as a result of clonal expansion. This process could also involve inactivation of a gene and there may be multiple genes involved.
There are several candidate genes for oesophageal metaplasia, although their possible involvement is mainly derived from evidence in other organ systems. The discussion here is restricted to some evidence-based examples.
p63 transcription factor
p63 is a homologue of the tumour suppressor and transcription factor p53. p63 is normally absent in simple, columnar epithelia, but is expressed in the basal layer of squamous epithelia. Recently, it has been shown that p63 is essential during normal oesophageal development. p63 knockout mice developed highly ordered, columnar, ciliated oesophageal epithelium.31
Homeobox genes Cdx1/2
The homeobox or HOX family of genes are developmental transcription factors. Various evidence suggests that Cdx2 is a “master switch” gene whose normal expression determines the proximal and distal specialisation of the gut in embryogenesis.32 In adulthood, Cdx1/2 protein is predominantly expressed in the small intestine and colon, but not in the stomach or oesophagus.33 Transgenic mice overexpressing Cdx2 develop intestinal metaplasia in the gastric epithelium34 and, conversely, the loss of Cdx2 leads to the development of polyp-like lesions in the intestine containing areas of squamous epithelium. Interestingly, the oesophagus-like squamous lesions are flanked by heterotopic gastric epithelium.35 This is a process called intercalary regeneration, by which tissue regeneration occurs at a junction between experimentally produced body parts to “fill in” those parts that normally lie between them. With regard to Barrett’s metaplasia, Cdx2 expression is observed in areas of intestinal metaplasia.36–38 Gastric metaplasia, which is also commonly observed in Barrett’s oesophagus, could be a form of intercalary regeneration. This is in contrast with the theory that gastric-type metaplasia develops before the intestinal type.39
Extracellular matrix components
To achieve asymmetrical stem cell divisions of the oesophageal epithelium, it is necessary to reconstitute the oesophageal squamous keratinocytes on denuded connective tissue containing oesophageal laminin 2. Oesophageal keratinocytes cultured on connective tissue from the skin failed to show the expected pattern of differentiation.21 Furthermore, conversion of embryonic stem cells to columnar or squamous epithelia has been shown to depend on the components of the extracellular matrix.40
Cytokines and transforming growth factor β
In GORD, the supporting oesophageal stroma becomes infiltrated with inflammatory cells, and it has been shown that the cytokine profile of Barrett’s oesophagus is fundamentally different from oesophagitis.41,42 Barrett’s oesophagus has increased levels of T helper 2 (anti-inflammatory) cytokines and a reduction in the ability to signal through the transforming growth factor β (TGFβ) cascade owing to a decrease in the expression of the signalling components TβRII, Smad2 and Smad4.43 Whether this change in TGFβ signalling contributes to the induction of Barrett’s oesophagus or arises as a consequence of its development is unclear; however, several lines of evidence support a causative role for reduced TGFβ. Targeted loss of TGFβ signalling components in mice has shown that this pathway has an important role in the development of various organs and tissues such as heart, bone and vasculature.44 Thrombospondin 1 knockout mice express low levels of active TGFβ and have altered cell differentiation in their distal oesophagus,45 amounting to focal areas of columnar epithelium. We could speculate how according to the model of metaplasia discussed above29 (fig 3), a reduction in TGFβ signal propagation could decrease the expression of factors necessary for maintaining squamous differentiation, resulting in the formation of focal areas of Barrett’s oesophagus.
Mechanisms for change in gene activation state
The triggering mechanisms by which specific genes are activated or silenced, leading to the development of Barrett’s metaplasia, is not understood. There are many levels of control and these may be direct genetic (eg, mutations in specific genes) or epigenetic (non-sequence-based changes that are inherited through cell division) modifications.
Examples of epigenetic silencing include the addition of methyl groups to the Cdx1 promoter.46 The transdifferentiation process in embryonic mouse oesophagus, discussed earlier, occurs by methylation of the keratin 8 promoter.20 Loss of TGFβ signalling occurs owing to several different mechanisms, including methylation, gene deletion and protein modification.43
In view of the role of GORD in the pathogenesis of Barrett’s oesophagus, it is interesting to know how this may directly affect gene expression. The gastroduodenal refluxate is a complex mixture of bile salts and dietary components at variable pH. Whether the refluxate is genotoxic will depend on its specific constituents. However, even those refluxate components that are not directly genotoxic47 may have profound effects on cell signalling and induce epigenetic effects on postmitotic cells. For example, nuclear factor κB may be activated by components of refluxate and inflammatory cytokines, which may in turn affect the expression of Cdx1/2.46,48 Chronic acid exposure has been shown to induce Cdx2 expression in primary mouse squamous oesophageal cells.49 This, coupled with the observation that Cdx2 mRNA is expressed in oesophagitis before the induction of Cdx1 or other intestine-specific genes,36 offers a possible link between gastro-oesophageal reflux exposure and the molecular basis for metaplasia. Mitogen-activated protein kinase (MAPK) signalling pathways can also be activated by gastro-oesophageal reflux exposure,50 and variations in integrin expression are known to regulate cell differentiation via MAPK signalling.51 Hence, through multiple cell signalling pathways, exposure to refluxate may have effects on cells that are removed from the site of the original stimulus. This may explain why the stem cell niche could be affected by exposure to refluxate even in its basal interpapillary location distant from the luminal surface (box 1).
Box 1: Induction of Barrett’s metaplasia
-
Luminal factors such as reflux components might be involved in causation.
-
Barrett’s metaplasia could arise from
transdifferentiation of a mature squamous cell,
tissue-specific or bone marrow-derived pluripotential stem cell.
-
Multiple levels of control over cell fate
cell signalling pathways,
genetic and epigenetic effects on cell-activation status.
-
Luminal environment can exert effects on cells distant to stimulus via cell signalling pathways.
MALIGNANT TRANSFORMATION
The progression of Barrett’s oesophagus to adenocarcinoma is generally thought to occur when a single Barrett’s epithelial cell is transformed into a cancer-initiating cell (cancer stem cell) through a series of genetic changes. The cancer stem cell or progenitor cell is a relatively recent term, coined because of its capacity for self-renewal. When the environment is conducive to multiple rounds of cell division, this may lead to malignant growth.
In the development of cancer, the occurrence of sequential genetic changes are responsible for clonal selection and tumour heterogeneity. The age-dependent incidence of many cancers including oesophageal cancer implicates four to seven rate-limiting, stochastic events.52 It is now becoming clear that although the nature and timing of these genetic events may be extremely variable, for an invasive carcinoma to develop, the specific genetic changes are not as important as the acquisition of certain biological capabilities. Six essential steps have been described for the development of cancer: self-sufficiency in growth, evading apoptosis, insensitivity to anti-growth, limitless replicative potential, sustained angiogenesis, and ability for invasion and metastasis.53 In Barrett’s carcinogenesis there is clear documentation for all of these biological characteristics, and the causative genetic changes may be as subtle as point mutations and as obvious as changes in chromosome complement.54,55 This discussion will include possible mechanisms for the acquisition of cancer-prone properties rather than presenting an exhaustive catalogue of the myriad genetic changes that have been the subject of other reviews (box 2).56,57
Box 2: Malignant transformation of Barrett’s metaplasia
-
Sequential genetic changes confer biological advantage.
-
Changes include genetic, epigenetic and gross chromosomal changes.
-
Biological characteristics acquired include abnormal growth control and increased invasion.
-
Chronic reflux exposure may contribute to pro-proliferative drive.
Abnormal growth
The first three biological capabilities are all related to abnormal growth. In Barrett’s carcinogenesis, this can be clearly seen in the increased expression of proliferation markers such as mini-chromosome maintenance proteins, with a shift in the proliferative compartment towards the surface.58–60 These proliferation abnormalities predate the development of dysplasia58 and are in keeping with the observed increase in the S phase (or DNA synthesis phase) of the cell cycle, which is an independent predictor of progression to cancer in Barrett’s oesophagus.61 Interestingly, despite early loss of the p16 gene,62,63 which controls the G2/S transition of the cell cycle, there does not seem to be an intrinsic abnormality in cell-cycle stage in Barrett’s carcinogenesis.60 This is in keeping with recent data obtained from gene expression profiling (whereby the expression of a large number of genes across the entire genome can be assayed simultaneously using microarray technology) in which there was no clear evidence that differential genes involved in cell-cycle regulation contributed to increased cell proliferation in Barrett’s carcinogenesis.64 This suggests that abnormal cell cycle entry or exit and possibly a shortened cell cycle length may be responsible for the increased proliferative index.
This general increase in proliferation would be in keeping with the idea that the micro-environment, including chronic, pulsatile exposure to luminal factors, is the pro-proliferative driver50,65 (fig 4). The acid-induced hyperproliferation appears to be dependent on activation of the Na+/H+ exchangers, that regulate intracellular pH in Barrett’s oesophageal cells, which in turn activate the p38 MAPK pathway to turn on mitogenic and antiapoptotic transcription factors.66,67 In addition, activation of the Na+/H+ exchanger leads to transient cytoplasmic alkalinisation,66 which is crucial for cells to progress from the G1 to S phase of the cell cycle.68 By using a similar in vitro model, it has since been shown that pulsatile bile exposure can also induce hyperproliferation via effects on protein kinase C.69 Repetitive exposure to GORD will also induce inflammation and hence changes in growth factors and cytokines. TGFβ is a good example of an immunoregulatory pathway that is commonly disrupted in Barrett’s carcinogenesis, leading to insensitivity to growth signals.70 The lack of TGFβ responsiveness is associated with a profound and progressive reduction in Smad4 that correlates with progression through the metaplasia–dysplasia–adenocarcinoma sequence.43
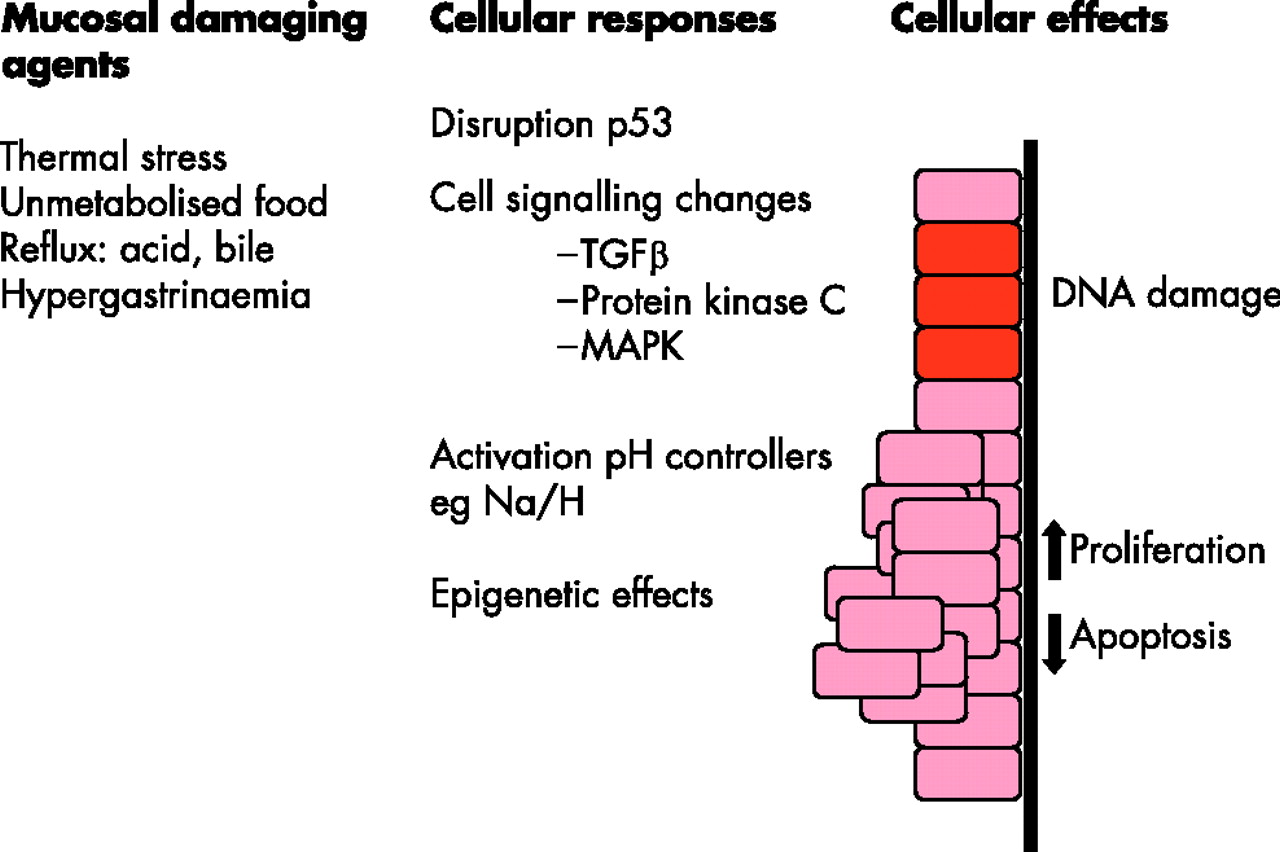
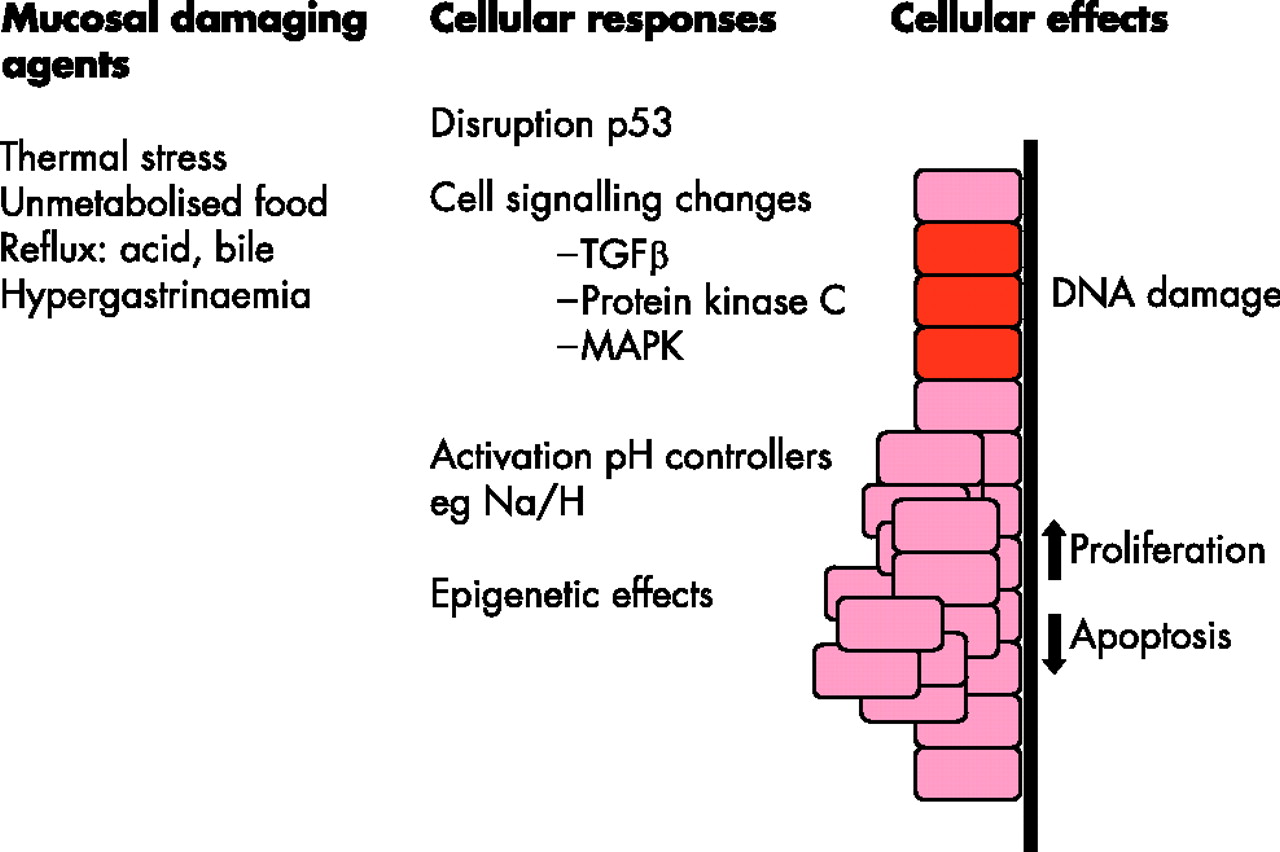
A summary of the pathways by which various mucosal damaging agents (left-hand column) may lead to changes in specific genes and cell signalling pathways (middle column) and hence to a variety of cellular effects (right-hand column), which may increase the propensity for cancer to develop. TGFβ, transforming growth factor β; MAPK, mitogen-activated protein kinase.
Overall, the persistent proliferative drive through exposure to luminal factors may explain the relative paucity of key oncogenes causally implicated in Barrett’s adenocarcinoma.
Evading apoptosis
Tissue growth is determined by the balance between cell death and proliferation. Reduced programmed cell death or apoptosis has been well-documented in many cancers, including Barrett’s carcinogenesis.70,71 Several genes are involved in controlling apoptosis, and expression profiling has shown that several of these are down regulated in the progression from Barrett’s oesophageal epithelium to cancer,64 including survivin and caspases.72–74 The microenvironment is an important determinant for the apoptotic index in a similar way to that discussed in the context of proliferation. In vitro exposure of an oesophageal adenocarcinoma cell line to acid led to the immediate down regulation of genes associated with apoptosis and early up regulation of genes associated with proliferation.67 The gene expression profiles suggest that MAPK pathways may be involved and suppression of apoptosis may occur via p53-dependent mechanisms.67 Decreased expression of Fas results in resistance to Fas-mediated apoptosis in oesophageal adenocarcinoma.75 The mechanism for this seems to be mediated via the Src kinase Yes, which is an upstream target of bile acids.76 The gastrin CCK2R receptor can also affect transcription of Fas via the phosphorylation of protein kinase B (PKB/Akt) and in vitro evidence suggests that gastrin may aid progression of antiapoptotic pathways via these signalling mechanisms.77 This may be relevant for patients with hypergastrinaemia secondary to proton pump inhibitors (PPIs).
Angiogenesis, invasion and metastasis
Angiogenesis is required to maintain tumour growth and to facilitate invasion and vascular spread. Vascular endothelial growth factor is a well-described determinant of new vessel formation, and this growth factor is expressed by the Barrett’s epithelial goblet cells and by the immature blood vessels that develop before the development of invasive adenocarcinoma.78,79 The cyclooxygenase 2 (COX2) enzyme also has a role in inducing angiogenesis80 as well as effects on immune surveillance, tissue growth and cell adhesion. The role of COX2 in the progression of Barrett’s oesophagus is somewhat conflicting.81–83 In a longitudinal case–control study, the COX2 expression levels in patients increased over time, regardless of the degree of malignant progression, and were independent of the tumour differentiation status or the degree of inflammation.84 Acid and bile have been shown to increase COX2 expression85, and interestingly, gastrin markedly induced COX2, prostaglandin E(2) and cell proliferation in biopsy specimens and cell lines that could be inhibited by CCK2R antagonists.84 Hence, the increase in COX2 expression could be occurring secondary to PPI use.
Wnt glycoproteins comprise a family of extracellular signalling ligands that have essential roles in the regulation of cell growth, motility and differentiation. Mutations in Wnt signalling molecules are carcinogenic through activation of β-catenin-TCF (T cell factor/lymphocyte enhancer factor) signalling. Previous studies86 have reported that nuclear accumulation of β-catenin is an indicator of activation of Wnt/β-catenin signalling, and this nuclear accumulation has been found in Barrett’s carcinogenesis. However, as mutations in adenomatous polyposis coli (APC) are rare, the mechanism of Wnt pathway activation has not been clear. Recent evidence suggests that the APC and SFRP1 (secreted frizzled-related protein) genes are inactivated by promoter methylation.87 However, as the loss of 5q and methylation of APC can occur both before and after the emergence of oesophageal adenocarcinoma, this suggests that the loss of APC is not necessary for cancer progression. This highlights the fact that during the genetic evolution of Barrett’s cancer there will be “hitchhiker lesions”. In other words, non-causative genetic lesions may occur and spread over large areas of the Barrett’s oesophageal mucosa if they coexist with a selectively advantageous lesion that undergoes clonal expansion.88 Therefore, not all genetic changes identified will have a causative role unless they confer a biological advantage on cell behaviour.
Genetic instability
The propensity for these six tumorigenic steps to occur is increased by underlying genetic instability. Reflux exposure has been shown to cause non-specific DNA damage,89 and the most prominent gene abnormality that promotes mutagenesis in response to DNA damage is the loss of the p53 tumour suppressor protein. When DNA damage occurs and p53 is functioning correctly, it leads to cell-cycle arrest to allow for DNA repair or apoptosis if the damage is excessive. Hence, in a normal cell, there are enough biological checkpoints in place to prevent premalignant cells progressing towards cancer (fig 5). In most, if not all human cancers, the p53 DNA damage signalling pathway is lost,90 and Barrett’s adenocarcinoma is no exception.55,91–95 Reid et al have shown that p53 loss most commonly occurs via mutation followed by loss of a chromosomal region, an event called loss of heterozygosity. Loss of p53 gene expression can occupy large areas of the Barrett’s oesophageal mucosa via clonal expansion.95 A proteomics approach also identified up regulation of an oestrogen receptor-responsive protein called anterior gradient-2 in Barrett’s oesophagus as an alternative way of silencing the p53 transcriptional response to DNA damage.96 In addition, there is evidence that environmental agents, such as a low oxygen concentration, low extracellular pH and thermal injury, can activate p53 protein and play a part in tissue transformation.97,98

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The chronic injury imposed on the oesophageal epithelium as a result of gastrooesophageal reflux may result in non-specific DNA damage, resulting in an increased propensity for further genetic abnormalities predisposing to cancer. Abnormalities in the tumour suppressor gene p53 are common in the progression of Barrett’s oesophagus. Changes in this pathway will prevent the cellular checking mechanisms, that normally prevent the perpetuation of genetic errors (green type depicts the normal cellular response and red type the abnormal response). GORD, gastro-oesophageal reflux disease.
Epigenetic changes are another causative mechanism for genomic instability. These may be global changes such as hypomethylation or hypermethylation of DNA, changes in the histones which make up the chromatin, as well as gene-specific effects. Various lines of evidence have led to the hypothesis that epigenetic changes may occur early in cancer development, leading to a polyclonal precursor of “neoplasia-ready” cells susceptible to environmental and age-dependent damage. Later, specific classical genetic changes in oncogenes and tumour-suppressor genes rendered vulnerable as a result of epigenetic modification can lead to monoclonal expansion.99 In Barrett’s carcinogenesis, changes in methylation status have been identified across several genes.100,101 These early epigenetic changes could arise as a result of chronic injury—for example, through reflux exposure, and would again help to explain why such exposures could be cancer promoting even when they are not inherently mutagenic. This hypothesis has not been specifically tested in the context of Barrett’s adenocarcinoma; however, liver regeneration after tissue injury leads to widespread hypomethylation,102 and environmental stress titrates out HSP90, which is required to fold the SMYD3 histone H3-K4 methyltransferase.103
Chromosomal instability is another form of genetic instability and changes in microsatellite allele sizes (DNA made up of short repetitive sequences) have been commonly observed in Barrett’s oesophagus,88,104 although not at a frequency that qualifies them as “microsatellite unstable” (replication slippage occurring as a result of microsatellites as seen in hereditary non-polyposis coli cancer). During tumour progression, widespread chromosomal losses and gains occur,105 and aneuploidy (deviation from normal diploid chromosomal number) is commonly observed late in progression.88 Whether altered chromosomal copy number represents a primary event or whether this is a secondary event occurring with uncontrolled proliferation is a question of debate and ongoing research.106
CLINICAL RAMIFICATIONS (BOX 3)
Box 3: Clinical developments
-
Biomarker discovery needs to be coupled with clinical assay development.
-
Risk stratification using biomarkers will help determine individual patient management, such as ablation treatment, chemoprevention or molecular-targeted treatments.
-
Chemoprevention requires testing in large, prospective randomised controlled trials.
Biomarkers
Part of the impetus for elucidating the molecular changes occurring throughout the metaplasia–dysplasia–carcinoma sequence is to identify molecular changes or biomarkers predictive for cancer development. The most promising data come from the combination of p53 and aneuploidy status from flow cytometric studies, which have shown a relative risk of 4 for progression to adenocarcinoma when a clone containing both of these abnormalities is >5 cm.88 Unfortunately, despite the identification of several molecular changes, none of these has yet been adopted in routine practice. Part of the reason for this is the paucity of molecular changes that have been validated in phase 4 studies (prospective studies that have been evaluated in large patient cohorts).107 The other reason for the lack of clinically useful biomarkers may result from the fact that cancer progression is non-linear and results from global genetic instability and epigenetic changes. Hence, future efforts to define biomarkers may be better focused on assaying for the acquired biological properties that occur during cancer development rather than focusing on specific genes. For example, there is an increase in the proliferation marker mini-chromosome maintenance 2 expression during the malignant progression of Barrett’s oesophagus. Furthermore, in a longitudinal case–control study, mini-chromosome maintenance 2 expression was increased before the development of dysplasia in the patients who went on to develop cancer.58 It is also hoped that new biomarkers will emerge from the systematic evaluation of changes in expression and epigenetic changes, such as methylation, across large numbers of genes.87,108,109 In the future, a systematic approach will be required to define sensitive and specific markers that are measurable using robust assays applicable to routine clinical practice.
Tailored therapy and chemoprevention
In the cancer specialty, marked advances have been made in identifying specific treatments as a result of knowledge of critical genetic changes, such as HER2/NEU amplification in breast cancer and ERBB2 mutations in lung cancer. However, in view of the complexity and non-linear pathways for cancer development and progression, it is becoming clear that only a subset of patients with a given tumour type will benefit from these treatments.110 Therefore, as more targeted treatments become available they will probably need to be tailored to the molecular basis of an individual’s tumour. In Barrett’s oesophagus, there are several different strategies that could be taken—for example, targeting of cell signalling pathways important in the disease pathogenesis, such as MAPK, nuclear factor-κB and COX 2. In addition, if it is proved that epigenetic changes are fundamental to the early stages of Barrett’s carcinogenesis, then agents that modify the epigenome globally, such as 5-aza-2′-deoxycytidine, which inhibits DNA methylation, may prove useful, and ultimately we may be able to target specific epigenetic modifications.111 However, the possible adverse consequences of such approaches should also be borne in mind. For example, global hypomethylation induced by 5-aza-2′-deoxycytidine could be detrimental for transcriptional regulation. These therapeutic modalities are at the preclinical stage in Barrett’s oesophagus.
To justify the treatment of patients with premalignant Barrett’s oesophagus, which has an overall low rate of malignant conversion, it will be necessary to target the patients at highest risk for developing cancer, or else to use treatments that are very safe and confer general health benefits. To date, there has been considerable interest in the use of acid suppressants and non-steroidal anti-inflammatory drugs. The rationale for the use of acid suppression arises from the in vitro and ex vivo data discussed above, which suggest that acid may adversely affect cell kinetics, activate COX2 and MAPK pathways, and cause DNA damage. Thus far, the data on whether acid suppression is a useful chemopreventive strategy are conflicting.112–114 The use of varying acid-suppression regimens and the lack of data from large prospective randomised trials make it difficult to evaluate their role. Laboratory and epidemiological data suggest that aspirin and non-steroidal anti-inflammatory drugs may be chemopreventive through their inhibitory effect on COX2.115–118 A prospective study has recently suggested that people who took aspirin or non-steroidal anti-inflammatory drugs regularly had a substantially lower incidence of oesophageal adenocarcinoma and aneuploidy than those who did not take such drugs regularly.119 The AspECT chemoprevention trial in the UK is a large randomised, prospective trial that aims to discuss the role of high-dose versus low-dose esomeprazole (PPI) with low dose or no aspirin on the overall mortality of patients with Barrett’s oesophagus.
CONCLUSIONS
Much work still needs to be done to better characterise oesophageal stem cells in terms of molecular markers and to establish experimental systems in which the control of stem cell behaviour and transdifferentiation can be investigated. It is becoming clear that there are multiple layers of control over cell fate, and that the microenvironment is important. Hence, gastro-oesophageal refluxate seems to have several effects at the molecular level both in terms of the development and progression of Barrett’s oesophagus. The refluxate may cause non-specific DNA damage as well as being a pro-proliferative drive in the context of genetic instability. This may set the stage for the non-linear acquisition of biological properties conducive to cancer development. Therefore, a different conceptual framework is required, which embraces the multiple pathways to Barrett’s-associated cancer so that clinical methods to identify high-risk people and to provide treatment take account of this individual variation in genetic changes.