Article Text

Statistics from Altmetric.com
A taxia-telangiectasia (A-T) is a multisystem autosomal recessive disorder, with an estimated frequency of 1/40 000-1/100 000 live births.1 The ataxia-telangiectasia mutated gene (ATM), located on chromosome 11q22-23,2 encodes a nuclear 370 kDa phosphoprotein, homologous to a family of phosphatidylinositol kinase related proteins involved in DNA damage response and cell cycle regulation.3–6 Classical A-T patients show progressive cerebellar degeneration with onset in childhood, oculocutaneous telangiectasia, variable immunodeficiency, recurrent sinopulmonary infections, and high levels of serum α-fetoprotein, chromosomal instability, and predisposition to lymphoid malignancies. The majority of patients are compound heterozygotes for two truncating ATM mutations with no detectable ATM protein. The A-T phenotype, therefore, is most commonly the result of null alleles, although missense mutations can also destabilise the protein, with similar consequences.7–9
Milder cases, designated A-T variants, are a heterogeneous group characterised by a combination of one or more of the following: later onset of clinical signs, slower progression, extended life span when compared to most classical A-T patients, and decreased levels of chromosomal instability and cellular radiosensitivity.10–12 In these patients telangiectasia and/or immunodeficiency can be absent while secondary features of A-T, such as peripheral neuropathy, dysarthria, chorea and/or dystonia, are present. Cancer and recurrent sinopulmonary infections may also be absent or reduced. The genotype of A-T variants is mostly compound heterozygous for a severe mutation together with a mild or leaky mutation, which is expected to express some ATM protein with residual function.13–15
A normal level of ATM protein in A-T variants can also be suggestive of mutations in other genes such as hMRE11, mutated in an A-T-like disorder with a classical A-T phenotype without telangiectasia.16–17 Other syndromes, such as Nijmegen breakage syndrome (NBS), include some A-T signs combined with a typical facies, sinopulmonary infections, microcephaly, progressive deterioration of intellectual function, early retardation of height growth, normal serum α-fetoprotein levels, and lack of telangiectasia18 (table 1). A-T and NBS are almost indistinguishable at the cellular level. The gene mutated in NBS is p95-NBS1 which encodes a component of the complex (p95/hMre11/hRad50) involved in DNA double strand break repair.19–20
Clinical and cellular phenotype in proband II.1 compared to A-T and NBS syndromes
Here we describe two sisters affected by a variant form of A-T with onset of ataxia at 27 years, showing polyneuropathy, choreoathetosis, and absence of telangiectasia, immunodeficiency, sinopulmonary infections, or cancer. Mutation screening of the ATM gene showed a compound heterozygous genotype with one missense mutation (8030 A→G in exon 57 which causes Y2677C) and a frameshift mutation (7481 ins A in exon 52). Neither mutation had previously been reported in A-T patients. Western blot analysis showed a low level of ATM protein which was able to phosphorylate p53ser15 significantly. We cannot exclude that neurological signs were present in II.1 before the age of 27 years but certainly these must have been very mild to have escaped medical observation. Therefore, patients II.1 and II.2 are among the mildest A-T variant phenotypes so far described. We suggest that the late onset of the disease and the milder phenotype can be explained by residual activity of the mutated protein.
CASE REPORTS
The proband (II.1, fig 1) was born to non-consanguineous parents originating from the same town in southern Italy. She was well until the age of 27 years, when she presented with unsteady gait and dyskinetic movements of the trunk, head, and limbs. At 42 years she was admitted for neurological assessment, with marked paretic gait and “stéppàge”, hypotrophy and hyposthenia of the four limbs, moderate dysarthria, chorea, and dystonia, and mild impairment of mental function (15% decrease with the Wechsler-Bellevue scale). Ankle and radial reflexes were absent, deep and shallow sensitivity was intact, and the cutaneous plantar reflex was normal. No alterations of the segmental cerebellum were evident and MRI of the brain showed moderate enlargement of subarachnoid spaces and normal cerebellum. EMG showed moderate signs of neurogenic impairment of the lower limbs. The patient did not show telangiectasia or abnormal eye movements, but an exudative maculopathy (right eye) with strabismus since childhood was noted. Haematological parameters were normal (IgM 1.41, IgA 3.35, and IgG 11.9 g/l), apart from serum α-fetoprotein, which was greatly increased (328.2 ng/ml, normal <10.9). Acanthocytes were absent and endocrinological assessment was in the normal range. The patient was given a generic diagnosis of “encephaloneuropathy of genetic origin”.

Pedigree of the family and linkage analysis at NBS and ATM loci. Haplotypes have been reconstructed from segregation analysis and show that affected sisters II.1 and II.2 share the same genotype A, B at the ATM locus.
The 44 year old sister (II.2) with a similar phenotype except for strabismus refused to undergo further diagnostic investigation. Both sisters have short stature (137 cm, <5th centile) compared with their sib. Sibs II.3, II.4, and II.5 and the father are in good health while the mother died from a stroke at 63 years. A female first cousin of the proband was affected by dyskinetic movements at 40 years and her brother was mentally retarded; their parents are the brother and the sister of the proband's father and mother, respectively.
Cytogenetic and molecular studies
Cytogenetic analysis was performed on peripheral blood lymphocytes of patient II.1. Cells were separated using Emagel (Hoechst, Germany), enriched with red cells, and incubated with phytohaemagglutinin (HA15, Murk, France) for 72 hours. Colcemid was added for the last two hours. Metaphase spreads were G-Wright banded and 27.4% of mitoses (23/84) showed translocations involving chromosomes 7 and 14. This finding of chromosomal instability is indicative both of A-T and NBS syndromes. The phenotype of our proband (II.1), while consistent with an A-T variant, also partially overlapped NBS for short stature and mental impairment. Moreover, a similar clinical phenotype with typical features of A-T and in addition microcephaly and mental retardation has also been described and named A-T-Fresno.21
Linkage of the patient's phenotype with the NBS locus (8q21) (markers D8S1811, D8S271, and D8S273, kindly provided by Professor C Sperling) and the ATM locus (11q23) (markers D11S2179 and D11S1818) was performed using fluorescently labelled primers with the ABI PRIM 377 sequencer and Genescan 2.1 software (Applera). This analysis excluded the NBS but not the ATM locus (fig 1).
Mutation screening of the ATM gene by PTT did not show any alteration. Each fragment was therefore sequenced and showed two variants (fig 2). One was a frameshift in exon 52 (7481insA) which produces a stop codon at position 7493 (codon 2498) causing protein instability and loss (fig 2A). Retrospectively, we noted that our PTT conditions had failed to detect this alteration in fragment 5, since the 66.7 kDa normal and the 61.4 kDa mutated protein were unresolved, and in fragment 6, since the 4.8 kDa protein had run out of the gel. The mutation was confirmed by direct sequencing of exon 52 from genomic DNA.

ATM gene mutation screening in II.1. (A) Genomic sequences of exons 52 (reverse primer) and 57. Mutation analysis of the ATM gene was initially performed by PTT and subsequently by cDNA sequencing. cDNA synthesis was performed under standard conditions.42 The entire coding region of the ATM gene was divided into six overlapping fragments: 1 (1634 bp, exons 4-13), 2 (1707 bp, exons 12-24), 3 (1668 bp, exons 22-32), 4 (1736 bp, exon 31-44), 5 (1820 bp, exons 41-53), 6 (1825 bp, exons 52-65). Forward primers were designed to include a T7 promoter sequence for the initiation of transcription by T7 RNA polymerase, as well as a eukaryotic translation-initiation sequence (T7: 5′-TAATACGACTCACTATAGGAACAG-ACCACCATC-specific primer 3′; specific primer sequences are available upon request). Protein truncation test was performed using the TnT coupled reticulocyte lysate system (Promega).43 Each fragment was gel purified (Qiaquick gel extraction kit, QIAGEN) and sequenced using internal primers and the Dye-terminator chemistry on a 377 ABI-Prism automatic sequence analyser (Applera). DNA from peripheral blood was extracted from all available sibs (II.1, II.2, II.3, II.4) using the Qiamp blood kit (QIAGEN). The 7481insA mutation was confirmed by genomic sequencing of exon 52; this was amplified using primers ex52f (5′-tcatgtgtgattttgtagttctgtta) and ex52r (5′-aagcacagggtagaatattggg) in standard conditions and the following cycling parameters: 94°C for three minutes, 35 cycles at 94°C for 30 seconds, 55°C for 30 seconds, 72° for 40 seconds, 72°C for 10 minutes. The 8030 A to G mutation was confirmed by allele specific oligonucleotide hybridisation (ASO) (primers 8030-A: 5′-aggagaatAtggaaatc and 8030-G: 5′-aggagaatGtggaaatc) after ex57 amplification in standard conditions (primers ex57f: 5′-aagtgcaaatagtgtatctgacc and ex57r: 5′-ttcatcactaaaactctaaggc)42 and the following cycling parameters: 94°C for three minutes, 35 cycles at 94°C for 30 seconds, 56°C for 30 seconds, 72°C for 40 seconds, and 72°C for 10 minutes. (B) Changes produced at nucleotide and amino acid level (codon numbering is consecutive from ATG). On the bottom a schematic representation of the two ATM mutated proteins is shown. Ficoll extracted peripheral blood lymphocytes from patient II.1 were transformed with Epstein-Barr virus to establish a lymphoblastoid cell line.
The second mutation was an A to G transition at nucleotide 8030 in exon 57, which changes a Tyr to a Cys at codon 2677 corresponding to a residue near the PI-3 kinase domain (fig 2B). Y2677C, confirmed at the genomic level, does not change ATM charge and hydrophobicity, and was excluded as a polymorphism because it was the only substitution observed in the complete coding sequence and it was not present in 100 control chromosomes screened by allele specific oligonucleotide hybridisation. The importance of Y2677 is also supported by its orthologous conservation. Neither mutation has been reported previously. Both mutations were confirmed in II.2 sharing the same genotype with II.1 and the 7481insA was also found in the heterozygous brother II.3.
DISCUSSION
To date, five missense mutations have been described in A-T variants: 7271T>G (V2424G in exon 51),14–22 8480T>G (F2827C in exon 60),14 7967T>C (L2656P in exon 56),23 6047A>G (D2016G in exon 43), and 1709T>C (F570S in exon 13)24; like Y2677C reported here, all reside outside the PI-3 kinase domain (aa 2857-2915). This could suggest that missense mutations outside the catalytic domain are associated with a residual ATM activity and result in the attenuated neurological signs observed in A-T variants.
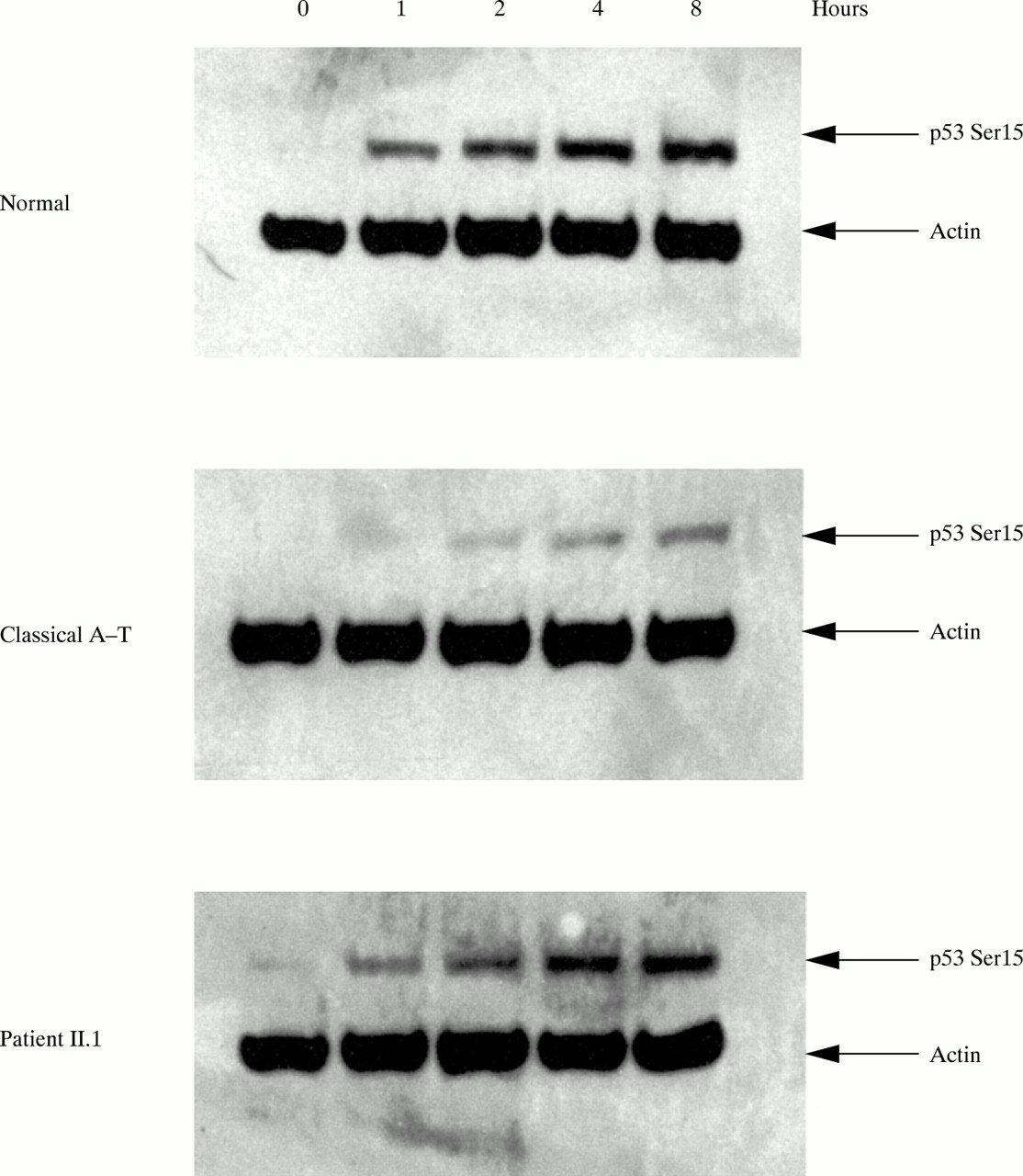
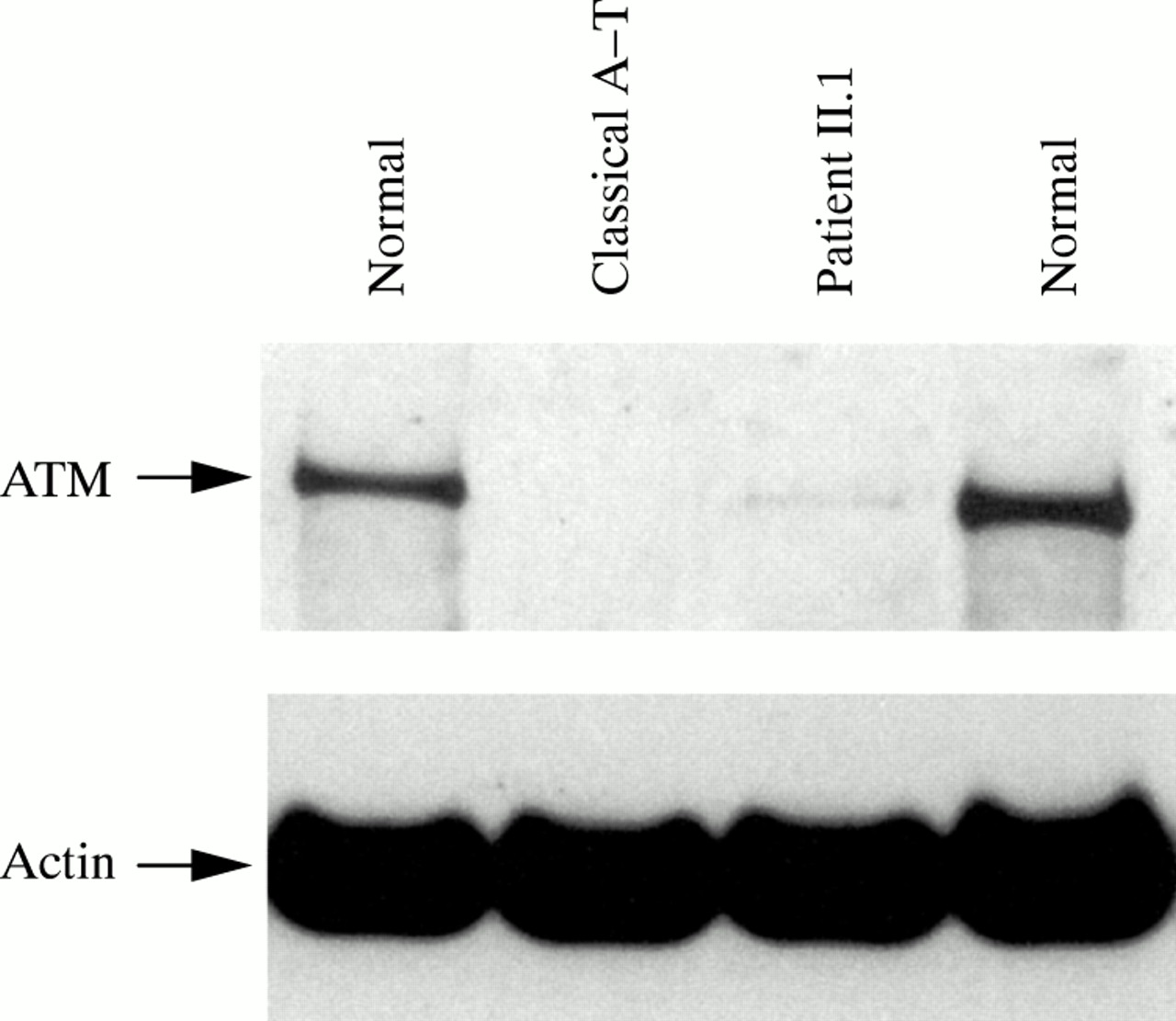
Western blot analysis on patient II.1 showed a low level of full length protein (<10%, fig 3) probably produced by the allele carrying the Y2677C missense mutation. These findings suggest that the milder phenotype observed in A-T variants and II.1 could be the result of some residual function of the ATM protein. Many of the functions of ATM occur via modulation of p53, which is phosphorylated in ser15 after ionising radiation exposure mainly by ATM and after a longer interval by its related kinase ATR.25–27 As a means of investigating the residual kinase activity of this mutant ATM protein, we assayed its ability to phosphorylate p53ser15 in the lymphoblastoid cells of II.1. The results showed a significant ability to phosphorylate p53ser15 at short times after irradiation, characteristic of phosphorylation by ATM, but less than is seen in normal cells (fig 4).

Western blot analysis of the ATM protein in patient II.1. Ficoll extracted peripheral blood lymphocytes from patient II.1 were transformed with Epstein-Barr virus to establish a lymphoblastoid cell line, and protein preparation and western blot were performed as described previously.15 ATM antibody recognises the N-terminal portion of the ATM protein (antibody FP822). Actin was used as a protein loading control. ATM is considerably reduced compared to controls (normal), but retains a minimal amount of full length product (<10%) as compared to a classical A-T patient.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
p53ser15 phosphorylation in an irradiated cell line of II.1. A residual p53ser15 phosphorylation is evident in cells derived from patient II.1 compared with a classical A-T patient and a normal subject, following exposure to ionising radiation. Cells were irradiated with 5 Gy of ionising radiation and harvested at one, two, four, and eight hours after irradiation. Whole cell lysate was separated on a SDS-PAGE gel and western blots were hybridised with p53 phosphoserine-15 (NEB) and actin.15
A major role of ATM is to regulate p53 responses which result in G1 arrest after exposure of the cell to some forms of DNA damage, particularly DNA double strand breaks caused by ionising radiation (IR). ATM also has role in the S phase and G2/M phase checkpoints following exposure of cells to IR.28–29 Activated ATM may exert its effect on p53 activity and stability by mediating simultaneous phosphorylation of both partners of the p53-MDM2 autoregulatory feedback loop and also Chk2.30 In A-T cells, where functional ATM is lost, the G1/S checkpoint is altered and cells accumulate genomic alterations. This instability could lead to a survival of damaged neuronal cells and to a selective neurodegeneration, as seen in cerebellar Purkinje and granular layer cells.31 The ability of the Y2677C protein to phosphorylate p53ser15 is an indication of retained kinase activity which may also allow some phosphorylation of other targets of ATM. The partial retention of p53ser15 phosphorylation in our patient might directly explain the mild symptoms and slow progression of the disease. However, while the stabilisation of p53 following ionising radiation is delayed in A-T cells, the kinetics and magnitude of their response in NBS and A-TLD cells are essentially unaffected,16–20 suggesting that ATM/p53 is not the sole pathway involved in cerebellar degeneration.
Since p53 loss is also critical to tumour development, retention of partial p53 phosphorylation could also explain the absence of lymphoid tumours in this variant. A functional link between ATM and p95/Nbs1 has been reported; after ionising radiation p95/Nbs1 is phosphorylated on ser343 in an ATM dependent manner in vitro and in vivo.32 It is noteworthy that p95 is part of a multiprotein complex hRad50/hMre11/Nbs1 involved in the DNA damage response to double strand breaks33 and hMRE11 is the gene mutated in A-TLD.16 The alteration of this pathway results in loss of the ionising radiation induced S phase checkpoint that is a common phenotype of both A-T and NBS cells.32–34 ATM is, however, a key protein involved in several networks as proven by the many reported interacting partners (BRCA1, c-Abl, p34, CtIP, β-adaptin, RPA).35–40 Therefore, other roles of ATM, such as alteration in cell membranes or cell signalling that depend on ATM, could be important and partially maintained in our patient. For instance, since both A-TLD and NBS patients do not show telangiectasia, the altered hRad50/hMre11/Nbs1 complex might not be responsible for this phenotype. The absence of this clinical feature in our patient suggests that the ATM dependent pathway leading to telangiectasia is not compromised.
A-T patients have very high levels of chromosome translocations in their peripheral lymphocytes involving breakage in the T cell receptor genes. This shows involvement of ATM in V(D)J recombination.41 The high frequency of t(7;14) translocations in this patient is interesting when compared with other milder cases of A-T, where the frequency of translocations tends to be lower than in classical A-T patients. The absence of a lymphoid tumour in these sibs may suggest some protection against such a tumour, especially T-PLL, but this is not certain and the presence of mutant protein may be an indication of risk of another tumour type.
Further molecular analyses of this A-T variant are needed to identify the pathways that regulate the severity of clinical symptoms. Their characterisation, also through expression profiles from microarray analysis, could help our understanding of the several functions of the ATM protein.
Acknowledgments
We thank Professor K Sperling (Institut für Humangenetik, Virchow Klinikum, Humboldt University, Berlin, Germany) for providing fluorescent markers on chromosome 8q21. The financial support of Fondazione Bertolini Torino and Fondazione E E Rulfo is gratefully acknowledged. We especially thank Professor M DeMarchi for discussion and critical reading of the manuscript. We would like to thank all family members for collaboration.