Article Text

Statistics from Altmetric.com
The role of the immune response in the control and eradication of leukaemia after allogeneic bone marrow transplantation is established but there is increasing evidence that the principal mechanism of cure after intensive chemotherapy alone is also immune mediated. The idea of a graft versus leukaemia (GvL) effect in eradicating leukaemia was suggested as early as 1957, when Barnes and colleagues1 showed in a murine model that leukaemic animals which received myeloablative radiotherapy followed by syngeneic bone marrow transplant all relapsed with the original acute leukaemia and died; in contrast, animals receiving the same radiotherapy but allogeneic bone marrow transplant all developed fatal graft-versus-host disease (GvHD). However, these latter animals survived longer than the recipients of syngeneic marrow and had no evidence of leukaemia relapse at necropsy. This provided evidence both that myeloablative radiotherapy does not eradicate leukaemia, and that an anti-leukaemia immune response was present. Immune mediated cure of residual leukaemia in the clinical setting has been suggested by observations of a reduced risk of leukaemia relapse after allogeneic bone marrow transplant when compared with autologous or syngeneic bone marrow transplant, and an association between graft-versus-host disease (GvHD) and disease-free survival.2,3 Furthermore, an increased incidence of leukaemia relapse has been reported after aggressive GvHD prophylaxis with cyclosporin or lymphocyte depletion. The most convincing direct evidence of allogeneic antileukaemia activity has been provided by the long lasting remissions following donor leucocyte infusions to treat relapse of chronic myeloid leukaemia (CML) after allogeneic stem cell transplantation.
Specific immunotherapy of acute myeloid leukaemia (AML) is the subject of intense research at present, with major research bodies such as the NIH, the Leukemia Society of America, and the Leukaemia Research Fund supporting large programmes in many centres. The approaches being taken differ widely between groups but can be most readily subdivided into those that rely upon an allogeneic transplant and those that aim to induce or maximise autologous immunity to residual disease.
Allogeneic responses
In allogeneic transplantation the aim is to maximise the GvL effect while minimising the risk and severity of GvHD. Post-transplant relapse of AML remains a problem, and subsequent survival rates have generally been very poor with either reinduction chemotherapy or second transplants. The advent of donor leucocyte infusions has provided an additional therapeutic option and the fact that complete remissions have been achieved is further “proof of principle” of the GvL effect of mature donor lymphocytes. Donor leucocyte infusions have been most effective in CML (76% complete remission), least in acute lymphoblastic leukaemia (ALL), with AML (29% complete remission) occupying the middle ground.4 In acute leukaemias, donor leucocyte infusions may need to be given in addition to prior chemotherapy.5 The full potential of donor leucocyte infusions thus far has been hindered by the concurrent risk of marrow aplasia (34%) and severe GvHD (41%).4 To minimise these risks, the optimal dose strategy for T cell addback has been investigated6 and, in relapsed CML, infusions are given cautiously, starting with doses of 105 lymphocytes/kg patient body weight and increasing over several weeks to doses of 1–5 107/kg in most cases. Eradication of the Philadelphia chromosome positive cells from the bone marrow of these patients takes many months and multiple lymphocyte infusions. Relapsed AML progresses too quickly for such a delayed response to be effective in most cases. Infusion of higher doses of donor leucocytes may resolve this, but the increased concomitant risk of severe or fatal GvHD is unacceptable.
The use of prophylactic donor leucocyte infusions is an approach being studied by several groups, with the intention both of treating undetectable residual disease and improving the immune reconstitution of recipients of T cell depleted allogeneic grafts. The timing of the infusions will be critical if GvL is to be retained without significant GvHD.7 Most groups are giving the first reinfusion around day 40 post-transplant but results are too preliminary to determine the effectiveness of this strategy in AML.
Given the aim of eradicating donor derived GvHD while retaining donor GvL and donor immunity to viral infections, some groups are trying to remove only the GvHD effectors from the graft ex vivo or in vivo. Ex vivo CD8 depletion8 has been tried with some success in CML but no results have been published from AML patients. Most recently, advances in clinical grade cell sorting have led to novel techniques for graft engineering, a field which is becoming known as “intelligent T cell depletion.”
Intelligent T cell depletion involves the removal of alloreactive cells which induce GvHD while preserving non-alloreactive cells. The ability to use HLA mismatched donors, ideally haplo-identical parents or children, will revolutionise allogeneic stem cell transplantation, as the greatest barrier to allogeneic stem cell transplantation remains the lack of suitable HLA identical donors. However, the severity of GvHD is unacceptable unless extreme (> 4 log) T cell depletion is employed. This severely delays immune reconstitution, leading to intense post-transplant immune suppression and deaths from relapses and disease.
Our group has looked at the activation marker CD69 in an ex vivo mixed lymphocyte culture of donor mononuclear cells and irradiated recipient cells as a tool to identify alloreactive GvHD inducing donor lymphocytes. Alloreactive cells can be efficiently depleted using magnetic beads, the allodepleted fraction retaining third party alloreactivity despite the removal of reactivity to the initial recipient cells.9 This mode of selective depletion only removes a small minority of donor cells and preserves a large pool of lymphocytes capable of anti-leukaemia activity, improving engraftment and hastening immune recovery. This system has been tested effectively in a mouse model of major histocompatibility complex (MHC) mismatched stem cell transplantation, with complete abrogation of clinical GvHD in the recipients of up to 2 × 107 donor lymphocytes. In contrast, 107 non-depleted donor cells were fatal in over 90% of animals (fig 1). Plans for a phase 1 clinical trial are under way.
Survival curve for NOD/SCID mice recipients of MHC mismatched donor splenocytes. Mice in the “non-depleted” group (n=11) received 107 non-manipulated donor splenocytes intraperitoneally; mice in the “depleted” group (n=12) received 107 donor splenocytes which had been depleted of alloreactive lymphocytes; “control” mice (n=8) received phosphate buffered saline alone.
Several other strategies have been used which either eliminate the alloreactive cells by immunotoxin10 or ricin11 conjugated to an anti-CD25 antibody, or alternatively anergise alloreactive cells by blocking the CD40:40 ligand12 or B7:CD2813 costimulatory pathways. The success of such strategies will rely upon the ability to dissect GvHD effector cells from those mediating GvL—that is, there must be leukaemia specific or leukaemia restricted responses. There is evidence supporting this assumption which is presented below. However, an alternate approach in which the GvHD response may be harnessed has been described by two European groups, and trials are under way: briefly, the donor lymphocytes in the allogeneic graft are transduced with a thymidine kinase (HSV-TK) suicide gene in a retroviral vector before infusion.14 In the event of clinical GvHD the transduced cells can be eliminated in vivo by administration of therapeutic doses of ganciclovir. The problems with the thymidine kinase transduced gene are that for the strategy to be successful, all alloreactive T cells must contain the gene, expression must be sustained in vivo, and the gene product must not be immunogenic. A further disadvantage is the loss of ganciclovir as a therapeutic option for CMV disease after transplantation.
With increased life expectancy, the numbers of elderly patients who may require allogeneic transplantation for AML are growing. The greatest advance for these patients will be the “mini-transplant” or “transplant lite.” This has sparked an explosion of interest in the last two years and aims to exploit immune mediated cure in transplantation, as patients do not receive myeloablative pretransplant conditioning.15,16 The purpose of pretransplant conditioning is to create a state of immunological host tolerance to the donor graft, allowing the induction of a graft versus leukaemia effect as the primary mode of treatment. As a state of tolerance is maintained, further infusions of donor lymphocytes can be given to maximise the GvL effect. Immune mediated treatment is especially attractive because less intensive conditioning is needed (hence, the term mini-transplant), which translates into decreased toxicity, improved immune reconstitution, avoidance of radiotherapy, and decreased incidence of GvHD, all of which are related to the intensity of conditioning.17
The safest allogeneic immunotherapy will rely upon the generation of non-alloreactive, donor derived, leukaemia specific or leukaemia restricted immunity. This has been hampered by the lack of known leukaemia specific tumour antigens. Tumour antigens promoting T cell responses have been studied mostly in the context of melanoma, but in AML they can either be alloreactive (MHC), tissue restricted (minor histocompatibility antigens, mHag),18 lineage and differentiation specific (proteinase 3),19 or leukaemia specific (PML-RARA in M3).20 In all the above, cytotoxic CD4 and CD8 T cells have been detected or generated which possess specific antileukaemia activity. CD8 clones specific for a variety of mHag can be isolated from most patients after transplantation, and some are selectively expressed on haematopoietic cells and so are suitable as a GvL target.
Professor Els Goulmy, at the University of Leiden, has been at the forefront of research into mHags for many years and was the first to isolate and sequence a human minor antigen. She has described two such antigens, HA-1 and HA-2, which are expressed exclusively on haematopoietic cells which are HLA-A2 positive.18 However, HLA-A2, HA-1, and HA-2 are not linked, so individuals who are HLA-A2 positive may not express HA-1 or HA-2 or may express one or both. Imagine a situation where both the donor and recipient are HLA-A2 matched (that is, around 70% of transplants in white patients) and the recipient is HA-1 positive. If the donor is HA-1 negative, the transplanted T cells will recognise the HLA-A2 molecules carrying the HA-1 mHag on the hosts myeloid cells as “foreign”; they will then generate a cytotoxic T cell response which selectively lyses only the recipient leukaemia and haematopoietic cells, while sparing donor haematopoietic cells and recipient fibroblasts, keratinocytes, and hepatocytes.21 The difficulty of finding donors who are HLA-A2 matched but HA-1 or HA-2 mismatched with the recipient may be overcome by the generation of specific T cell lines that are HLA-A2 restricted and are specific for each of the mHags to be used for treatment of relapse post-transplant, or for in vivo purging before transplantation. Minor antigens restricted to other HLA alleles will be identified if such an approach is successful in the HLA-A2 matched setting.
Specific fusion oncogenes such as PML/RAR in AML M320 or bcr/abl22 are potential tumour antigens, and various methods of generating cytotoxic T cells to these have been tried, including bulk cultures, limiting dilution assays, and dendritic cells.22–24 The emergence of tetramer technology, in which complexes of HLA molecules and peptides are manufactured artificially, allows us to visualise and select specific peptide restricted cytotoxic T cells for expansion and therapeutic application. This method has been successfully used to identify mHag specific cytotoxic T cells25 and a project supported by the LRF Immunotherapy Initiative is using these reagents to derive cytomegalovirus (CMV) specific cytotoxic T cells to treat CMV infection post-transplant (Moss P, personal communication).
The exciting possibility of using the catalytic subunit of telomerase as a universal tumour antigen has also recently been described.26 T cells generated against peptides of this tumour antigen can recognise and lyse a variety of tumours, including lymphoma and myeloma, in an HLA restricted manner.
Currently, allogeneic antileukaemia activity is recognised and some target antigens have been identified. Problems of donor availability remain acute and research into optimising the safety of multiply HLA mismatched transplants is under way. Rendering the mismatched transplants safe with respect to GvHD may abrogate GvL and thus the optimal treatment may involve combination treatment by passive immunotherapy with leukaemia restricted cytotoxic T cell lines.
Autologous responses
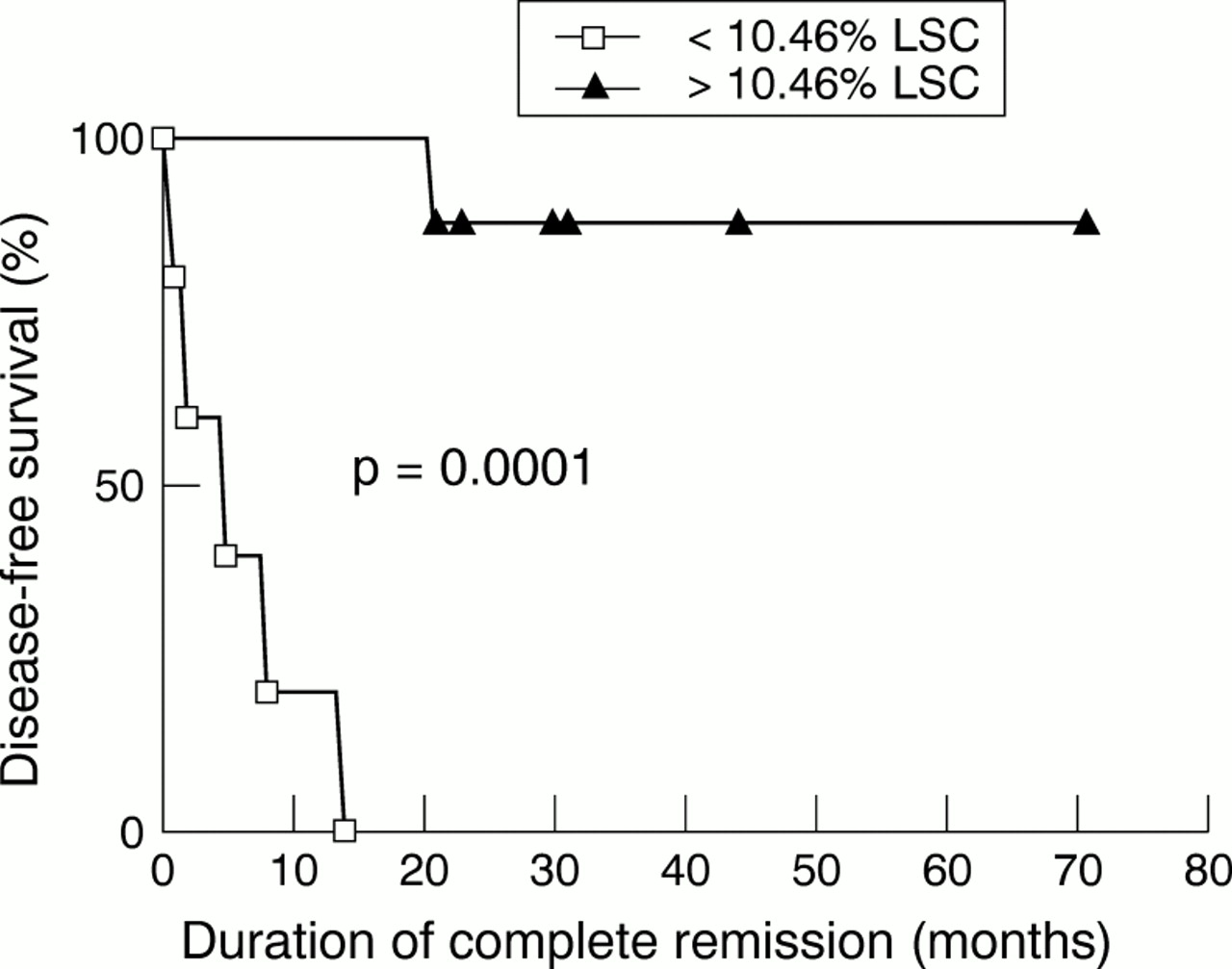
All patients who achieve complete remission after chemotherapy are potential recipients of autologous immunotherapy, with or without concomitant stem cell transplantation. This concept is not new: advent of recombinant interleukin-2 in the 1980s led to a number of clinical trials.27–30 The overall results—including those from large, randomised, but unreported studies—have been disappointing, and most trials in acute leukaemia have ceased. However, our understanding of the human immune system has advanced enormously since these early trials, and the field is both active and optimistic once more. Particularly encouraging is the evidence that patients can spontaneously develop immunity to residual leukaemia after chemotherapy31,32 and that this immunity can be measured in vitro and induced in some patients by administration of α interferon.33 In a recent study from our group, a cut-off level of leukaemia-specific cytotoxicity in peripheral blood samples of patients in remission was determined which predicted disease-free survival beyond two years with high specificity and sensitivity (fig 2). Furthermore, the cells which mediated this activity have been identified as a subset of natural killer (NK) cells and work on their expansion for passive immunotherapy is underway.

{kind=link}
{kind=link}
Disease-free survival of patients with acute leukaemia by virtue of the level of autologous leukaemia specific cytotoxicity measured in peripheral blood samples taken in remission after completion of treatment.
Passive immunotherapy with autologous natural killer cells is logistically attractive for many reasons, but for continued immunological memory a specific T cell response is required. Evidence for spontaneous T cell responses to AML is lacking but our current level of understanding of the nature of naive immune responses suggests that we will be able to generate such cells in vitro and or in vivo. Several strategies are being employed.
Dendritic cells are the most efficient antigen presenting cells and these can now be cultured from monocytes and from CD34 progenitors. Dendritic cells can be pulsed with peptides, proteins, or tumour lysates, a strategy that has already been used to feed leukaemia derived peptides (synthetic and eluted) into dendritic cells to obtain specific T cell effectors.22,34,35 Data were presented recently on the successful generation in vitro of specific cytotoxic T cell to autologous CLL cells using a dendritic cell strategy.36 Others have tried using these peptides and dendritic cells for in vivo vaccination strategies which may improve responses compared to vaccination with proteins, irradiated leukaemic blasts, or even immunogenic peptides alone.37 Tumour vaccines will probably work best in a minimal residual disease setting after chemotherapy. However, on many occasions, the tumour antigen may be unknown or it may not be possible to isolate and obtain exact tumour peptides. Moreover, predictions of MHC peptide specificity in vitro may not hold true in clinical practice. To bypass these problems of peptide generation, cytokines have been used to derive dendritic cells from autologous leukaemic blasts with the intention that these cells will present leukaemia restricted peptides innately.38 While this is a promising strategy, it is not applicable to all subtypes of AML. An alternative strategy is to fuse the tumour cell with the dendritic cell.39 This has been successful with carcinoma cells and could be extended into the context of AML. Also, the discovery that dendritic cells shed membrane vesicles called exosomes, which in themselves can function as efficient antigen presenting cells, has led to the pulsing of tumour peptides into exosomes.40
The other vital component in the generation of cytotoxic T cells is provided by CD40 on dendritic cells and CD40 ligand on CD4 cells.41 There has been a flurry of excitement recently in this field using activating anti-CD40 antibodies to modulate the function of dendritic cells. This augments responsiveness to tumour vaccination strategies as well as enhancing and priming the specific antitumour cytotoxic T cell response.42–44
AML blasts subvert the host immune surveillance by a myriad of mechanisms which include decreased expression of MHC molecules, secretion of immunosuppressive cytokines, a lack of costimulatory molecules,45,46 downregulation of CD3ζ, and tyrosine kinase expression.47 This results in the failure of “correct” antigen presentation to the T cell and hence a failure of the immune response. However, AML blasts can be genetically modified to improve their ability to present endogenous antigens.48 Our group has produced an adeno-associated virus (AAV) construct capable of simultaneously transducing primary human AML cells with genes encoding IL-12 and CD80, leading to subsequent protein synthesis and expression.49 There is marked synergism between IL-12 and CD8050 in the initiation of primary cytotoxic responses. We have termed these transduced cells “tAML” and have shown them to be immunostimulatory in vitro. We are currently using them to stimulate autologous peripheral blood mononuclear cells in vitro and expand reactive cells for analysis and in vitro testing. A passive immunotherapy programme with these reactive cytotoxic T cells is planned for patients in remission after chemotherapy. Ultimately, the tAML cells could be used as a vaccine in vivo to stimulate autologous immunity directly. This approach is being followed by the group at King's College Hospital in London where they are using an adenoviral vector to transduce AML blasts with CD80 and interleukin (IL)-2.
The immune system is regulated by a cytokine network and its importance in the pathophysiology of GvHD is well established.17 IL-2 increases antigen specific effector function and induces MHC unrestricted natural killer cell and cytotoxic T cell activity51 but, as discussed above, trials of IL-2 have been disappointing. Recently there has been a resurgence of interest52,53 and its use in combination with histamine has been proposed as the two are synergistic in cell mediated killing of leukaemic cells and in upregulation of CD3ζ chain expression.54
IL-12 is likely to prove a valuable immunotherapeutic cytokine. It drives reactive CD4 cells to a Th1 (cytotoxic) response55 and has been shown to promote GvL activity independent of GvHD. Despite this, systemic injection results in marked adverse effects and variable long term immunity; thus its successful clinical application will require more physiological delivery systems.
There has been extensive use of α interferon in CML but less so in AML. As described above, we have used it as maintenance therapy in AML, where it has been associated with induction of leukaemia specific cytotoxicity and long term disease-free survival.
Conventional chemotherapy, bone marrow transplantation, or systemic immunotherapy with cytokines are all associated with varying degrees of toxicity to other tissues. In contrast, targeted immunotherapy involves the specific delivery of either toxins, chemotherapy, or radioactive elements specifically to the leukaemia blasts, while avoiding toxicity to other organ systems. This can be done by identifying unique cell surface markers on leukaemia blasts like CD33 (present in 90% of AML blasts), and conjugating specific antibodies to a toxin/radionuclide for specific killing. Selective killing of AML blasts was achieved in a recently published phase I study using anti-CD33-calicheamicin immunoconjugate.56 Side effects were tolerable: postinfusion symptoms of fever, chills, and hypotension, as well as neutropenia. There was 20% success in this group of relapsed or refractory patients, although nearly all subsequently relapsed. Immunocytokines are another potential way forward, for example fusing IL-2 with a cell surface molecule or IL-4 with an immunotoxin for AML.34
AML targeted antibody treatments are not only restricted to cytotoxic effects. Another novel emerging mode of treatment is the use of anti-CD44 to induce differentiation in AML blasts, much as ATRA (ALL trans retinoic acid) is used with such success in the treatment of AML M3. A recent trial has produced promising initial results.57
Summary
Immunotherapy in the form of allogeneic GvL has been curing AML patients for nearly 30 years but our understanding of the mechanisms has been poor. Our rapidly evolving understanding of the human immune system and the concomitant technical developments in ex vivo cell manipulation, the vision of funding bodies, the dedication of clinical and research staff, and above all the commitment of our patients, promise substantial progress in the treatment of this disease in the year 2000 and beyond.
Acknowledgments
MBCK is a clinical training fellow of the Leukaemia Research Fund. Thanks to Dr Marion Wood for helpful comments regarding the structure of the manuscript.