Article Text

Abstract
Isocitrate dehydrogenase 1 (IDH1) encodes a protein which catalyses the oxidative decarboxylation of isocitrate to α-ketoglutarate. Mutant IDH1 favours the production of 2-hydroxyglutarate, an oncometabolite with multiple downstream effects which promote tumourigenesis. IDH1 mutations have been described in a number of neoplasms most notably low-grade diffuse gliomas, conventional central and periosteal cartilaginous tumours and cytogenetically normal acute myeloid leukaemia. Post zygotic somatic mutations of IDH1 characterise the majority of cases of Ollier disease and Maffucci syndrome. IDH1 mutations are uncommon in epithelial neoplasia but have been described in cholangiocarcinoma.
- genetics
- neoplasms
- molecular biology
Statistics from Altmetric.com
Introduction
Isocitrate dehydrogenases (IDHs) are enzymes that catalyse the oxidative decarboxylation of isocitrate to α-ketoglutarate.1 2 IDH exists in three isoforms in humans (IDH1, IDH2 and IDH3).3 IDH1 (cytosolic) and IDH2 (mitochondrial) are nicotinamide adenine dinucleotide phosphate (NADP+) dependent while IDH3 (mitochondrial) participates in the citric acid cycle and is nicotinamide adenine dinucleotide (NAD+) dependent. IDH1 and IDH2 are the isoforms implicated in tumourigenesis. Mutations in IDH2 occur in up to 40% of angioimmunoblastic T-cell lymphoma4 and occur more frequently in cytogenetically normal acute myeloid leukaemia (AML) than IDH1 mutations.5 IDH2 mutations are also implicated in a subset of chondrosarcoma,6 cholangiocarcinoma7 and low-grade diffuse gliomas.8 9 This review will focus on IDH1.
Structure
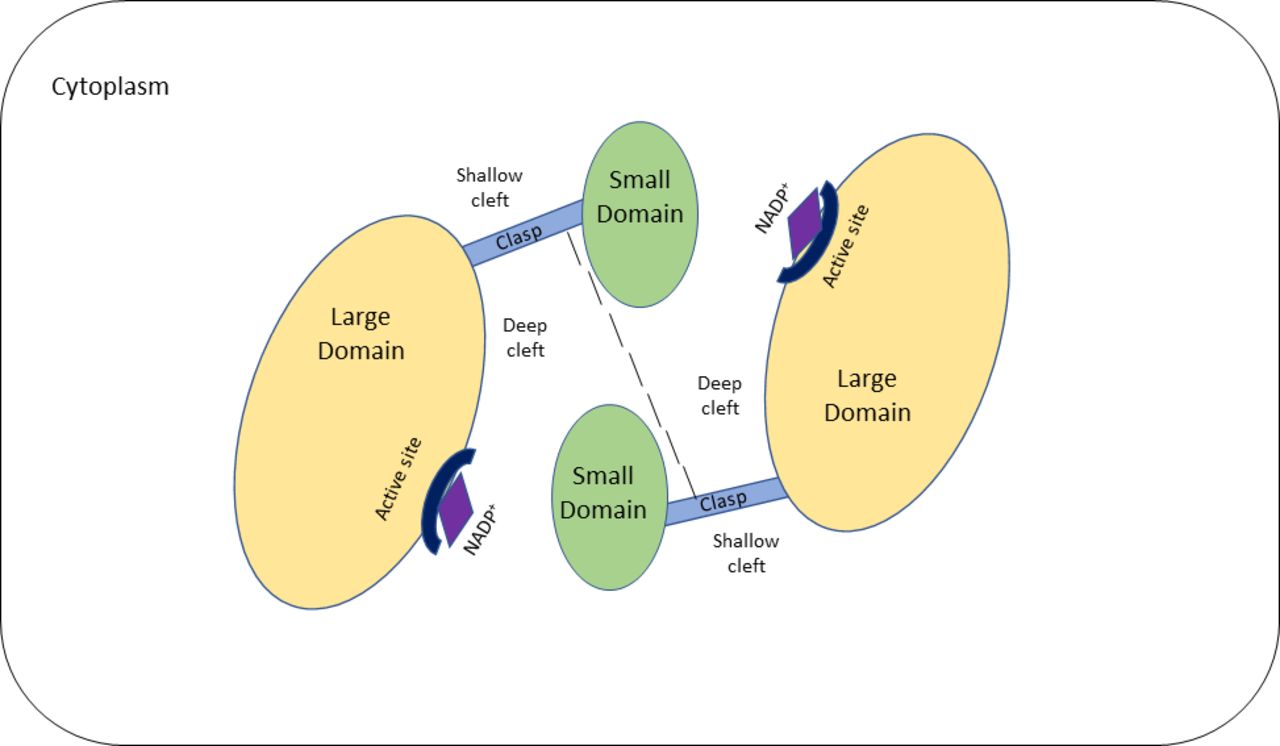
The IDH1 gene is located at chromosome 2q34 and contains 10 exons that span 18.9 kb.10 11 It encodes the IDH1 protein which comprises 414 amino acids with a molecular mass of 46.7 kD and is located within cytoplasm and peroxisomes.1 12 IDH1 is a homodimer comprising two hydrophilic active sites and two protein subunits or monomers.13 Each monomer is made up of a large domain, a small domain and a clasp domain with two clefts (figure 1).13 The deep cleft which lies between the large domain and small domain of one monomer and the small domain of the second monomer forms the active NADP+ binding site.13

Simplified schematic representation of isocitrate dehydrogenase (IDH1) in its open, inactive state. The IDH1 protein comprises two monomers which each consist of a large domain, small domain and clasp domain. Two clefts are present on either side of the clasp domain: a shallow and a deep cleft. The deep cleft lies between the large and small domain of one monomer and the small domain of the second monomer. The deep cleft contains the active binding site for NADP+. The two clasp regions join the two monomers together. NADP+, nicotinamide adenine dinucleotide phosphate.
Function
As discussed above, IDH1 catalyses the oxidative decarboxylation of isocitrate to α-ketoglutarate (figure 2).1 2 This process is NADP+ dependent and results in reduced nicotinamide adenine dinucleotide phosphate (NADPH).1 This reaction is reversible under physiological conditions.14 Production of cytoplasmic NADPH reduces intracellular oxidative stress while α-ketoglutarate maintains DNA and histone proteins in a demethylated state.13 15 16 IDH1 generates the NADPH required for cholesterol and fatty acid biosynthesis and plays a critical role in glucose-stimulated insulin secretion.11 17 Acetyl-coenzyme A synthesis from α-ketoglutarate under hypoxic conditions is required for lipogenesis.18

Schematic representation of the function of isocitrate dehydrogenase (IDH1). EgIN, Egl nine homolog 1; HIF, hypoxia-inducible factor; mIDH1, mutant IDH1; NAD, nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; wtIDH1, wild-type IDH1.
Mutant IDH1
IDH1 mutations are heterozygous oncogenic gain of function mutations resulting in one mutant and one wild-type allele.19 Recurrent missense mutations lead to a single amino-acid substitution of arginine at codon 132 in exon 4.19 The most common mutation variants include p.R132H and p.R132S although p.R132C, p.R132G and p.R132L have also been described.20 21 Mutation decreases the binding affinity of the active sites for isocitrate and increases their affinity for NADPH thereby impairing decarboxylation of isocitrate (the ‘forward’ reaction).14 The ‘reverse’ reaction is favoured but incomplete when IDH1 is mutated resulting in the production of the R-enantiomer of 2-hydroxyglutarate (2HG), an oncometabolite (figure 2).14 22 Furthermore, mutant IDH1 (mIDH1) production of (R)-2HG is enhanced by coexpression of wild-type IDH1 which provides the α-ketoglutarate substrate required for the reaction.14 23 Production of the R-enantiomer of 2HG has numerous downstream effects which promote tumourigenesis including inhibition of histone demethylation. The latter inhibits differentiation of progenitor cells and stimulates Egl nine homolog 1 which leads to diminished hypoxia-inducible factor.23 24 IDH1 mutation also results in a global DNA hypermethylator signature through inhibition of the ten-eleven translocation family and the Jumonji family of histone lysine demethylases.23–26
Gliomas
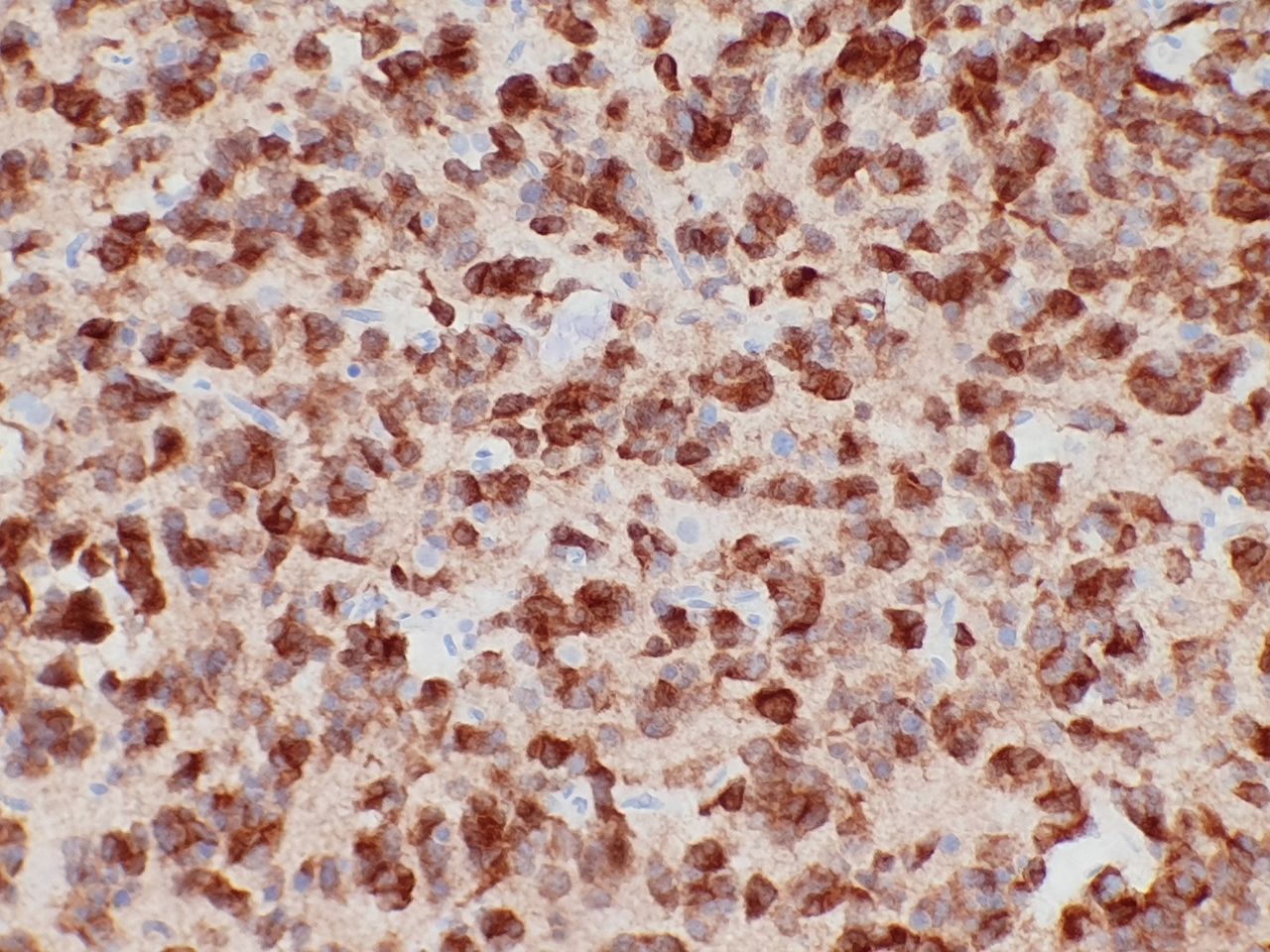
The overwhelming majority of WHO grade II or III diffuse gliomas (diffuse astrocytoma, anaplastic astrocytoma, oligodendroglioma, anaplastic oligodendroglioma) demonstrate IDH1 or IDH2 mutations.8 9 20 27 The majority of glioblastomas (WHO Grade IV) are IDH-wild type; however, those arising from low-grade gliomas (so-called secondary glioblastomas) are IDH mutant.9 27 28 In keeping with this, IDH-mutant glioblastomas occur in younger patients and demonstrate a better prognosis than wild-type tumours.20 The latest WHO classification of central nervous system tumours update of 2016 classifies glial tumours using an integrated genotype–phenotype approach based on the presence or absence of IDH mutation (IDH mutant or IDH wild type).29 The IDH1 R132H mutation is the most common in this setting (90% of IDH mutations) and an immunohistochemical stain against the mutant-specific antigen (R132H-mutant IDH1) has been developed (figure 3).30 This has become an essential tool in the workup of glial tumours and may also be used to distinguish a true glial neoplasm from reactive gliosis.31 32 Positive nuclear and cytoplasmic staining is seen in the majority of cells in IDH-mutant tumours.33 IDH1 R132H immunohistochemistry should be performed in all diffuse gliomas followed by IDH1 and IDH2 sequencing of all negative low-grade gliomas and glioblastomas in patients younger than 55 years.34
{kind=link}
{kind=link}
{kind=link}
Isocitrate dehydrogenase (IDH1) R132H immunohistochemical stain showing cytoplasmic and nuclear staining confirming an IDH1-mutant astrocytoma.
Cartilaginous neoplasms
Heterozygous IDH1 mutations have been demonstrated in 51% of conventional central and periosteal cartilaginous neoplasms including enchondroma, chondrosarcoma (grades 1–3), dedifferentiated chondrosarcoma, periosteal chondroma and periosteal chondrosarcoma.6 These mutations are absent in peripheral chondrosarcomas (associated with EXT1 and EXT2 mutations) and osteosarcoma.6 35 The presence of IDH1 mutation has been shown to be a useful tool to distinguish chondrosarcoma from chondroblastic osteosarcoma and dedifferentiated chondrosarcoma with osteosarcomatous differentiation from osteosarcoma.36
Enchondromatosis: Ollier disease and Maffucci syndrome
Enchondromatosis is a rare heterogenous disorder characterised by the presence of multiple symptomatic intramedullary cartilaginous neoplasms. Ollier disease and Maffucci syndrome are the most common subtypes and are typically non-familial disorders. Both disorders demonstrate multiple enchondromas involving the tubular bones of the limbs with an increased risk for development of secondary chondrosarcoma. Maffucci syndrome is characterised by the addition of soft tissue, visceral or cutaneous haemangiomas particularly spindle cell haemangiomas.37
IDH1 mutations have been described in the tumours of 85% of patients with Ollier disease and 81% of those with Maffucci syndrome.38 39 These postzygotic mutations are present in the enchondromas, chondrosarcomas and spindle cell haemangiomas of the afflicted with identical mutations identified in multiple tumour types from the same patient.38 39 Mutations described include R132C, R132H and R132G in exon 4.38 39 Maffucci syndrome shows exclusively R132C mutations. A low frequency of mIDH1 has been described in normal tissue from these patients.38
Spindle cell haemangioma
First described as spindle cell haemangioendothelioma, spindle cell haemangioma is now classified as a benign vascular neoplasm based on its excellent prognosis.40 41 When occurring outside the setting of multiple enchondromas, IDH1 mutations are seen in 64% of cases, a feature that has not been demonstrated in any other vascular lesions or malformations.42
Myeloid neoplasms
mIDH1 has been reported in AML (7%–14%),43 acute lymphoblastic leukaemia (5.5%),44 myelodysplastic syndromes (MDS; 3%)45 and myeloproliferative neoplasms (MPN). Most IDH1 mutations described in this setting involve a cysteine or histidine substitution for arginine at R132 (R132C or R132H). IDH2 mutations are more common than IDH1 mutations in AML and MDS.43 mIDH1 in AML is associated with cytogenetically normal AML, cytogenetically intermediate-risk AML and trisomy 8.43 Approximately 20% of MPN at leukaemic transformation show IDH1/2 mutations.46
The prognostic significance of IDH1 mutations in AML has been controversial.47 A large meta-analysis showed reduced overall survival and a lower rate of complete remission with cytotoxic chemotherapy.48 mIDH1 in MDS has a negative prognostic impact with reduced overall survival and higher rates of leukaemic transformation.49
Cholangiocarcinoma
Cholangiocarcinoma is a malignant tumour arising from biliary tract epithelium and can be classified as intrahepatic or extrahepatic based on anatomical location.50 The prognosis is poor with most patients demonstrating advanced disease at presentation.50 IDH1 mutations occur in 20%–35% of intrahepatic cholangiocarcinoma and only rarely in extrahepatic cholangiocarcinoma.7 51 Mutations described include R132C, R132L, R132G and R132S.7
Other neoplasms
Apart from cholangiocarcinoma, IDH1 mutations are rare in epithelial tumours but have been reported in a small subset of prostate adenocarcinoma (2.5%)52 and non-small cell lung carcinoma (0.6%).53 Novel IDH1 mutations have also been described in one case each of anaplastic thyroid carcinoma (G123R) and follicular thyroid cancer (V71I).54 Two of 39 malignant melanomas in one study showed IDH1 mutations, both of which occurred with either BRAF or KIT mutations.55 See table 1 for a summary of neoplasms frequently associated with IDH1 mutations.
Neoplasia frequently associated with IDH1 mutations
Targeted therapy
A number of strategies for targeted therapy in IDH1 mutant tumours have been investigated including hypomethylating agents, IDH mutant enzyme inhibitors, immunotherapy and BCL-2 inhibition. Preclinical studies have validated the proof of concept that targeted inhibition of IDH1 mutants results in decreased 2-HG, release of cellular differentiation block and reversal of histone and DNA hypermethylation.56 57 Four IDH1 inhibitors are currently under investigation in clinical trials for treatment of AML, gliomas and solid tumours. AG-120 (Ivosidenib, Tibsovo), an oral small-molecule inhibitor of mutant IDH1, was shown to have an acceptable safety profile when used as monotherapy for advanced solid tumours.58 AG-881 (Vorasidenib, an oral pan-mutant IDH1/2 inhibitor), BAY1436032 and DS-1001b (both IDH1 mutant inhibitors) are still under investigation to determine their safety profiles.59–61
Take home messages
Wild-type isocitrate dehydrogenase (IDH1) converts isocitrate to α-ketoglutarate. Mutant IDH1 converts α-ketoglutarate to the R-enantiomer of 2-hydroxyglutarate, an oncometabolite that results in tumourigenesis.
IDH1 mutations characterise low-grade diffuse glial neoplasms and are present in glioblastomas arising from low-grade gliomas. These patients have a better prognosis than IDH-wild type glioblastoma.
Nonfamilial postzygotic IDH1 mutations are present in the majority of patients with Ollier disease and Maffucci syndrome which results in multiple enchondromas and an increased risk of secondary chondrosarcoma.
Cytogenetically normal acute myeloid leukaemia and some other myeloid neoplasms demonstrate IDH1 mutations.
IDH1 mutations are also present in cholangiocarcinoma but are rare in other epithelial neoplasms.
References
Footnotes
Handling editor Runjan Chetty.
Contributors Both authors contributed equally.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; internally peer reviewed.