Article Text

Abstract
Squamous cell carcinoma of the head and neck (HNSCC) is a heterogeneous but largely preventable disease with complex molecular abnormalities. It arises from a premalignant progenitor followed by outgrowth of clonal populations associated with cumulative genetic alterations and phenotypic progression to invasive malignancy. These genetic alterations result in inactivation of multiple tumour suppressor genes and activation of proto-oncogenes, including p16ink4A, p53, cyclin D1, p14ARF, FHIT, RASSF1A, epidermal growth factor receptor (EGFR), and Rb. Intramucosal migration and clonal expansion of transformed cells with formation of abnormal genetic fields appear to be responsible for local recurrences and development of second primary tumours.
- CIS, carcinoma in situ
- HNSCC, squamous cell carcinoma of the head and neck
- HPV, human papillomavirus
- LOH, loss of heterozygosity
- SPT, second primary tumour
- HNSCC
- head and neck carcinoma
- papillomavirus
- molecular pathology
Statistics from Altmetric.com
- CIS, carcinoma in situ
- HNSCC, squamous cell carcinoma of the head and neck
- HPV, human papillomavirus
- LOH, loss of heterozygosity
- SPT, second primary tumour
Squamous cell carcinoma of the head and neck (HNSCC) remains a significant cause of morbidity and mortality, with approximately 540 000 new cases annually worldwide and 271 000 deaths for a mortality of 50%.1 An estimated 4350 new cases of oral and laryngeal carcinoma are diagnosed each year in Canada, with 1560 tumour related deaths giving a mortality of 36%.2 In the USA, the use of adjuvant radiotherapy after surgery, and concurrent chemotherapy and radiotherapy, has resulted in improved five year survival rates for advanced carcinomas of the larynx and pharynx but no survival improvement has been seen for oral carcinomas and early laryngeal carcinomas.3 Conventional HNSCC is largely a preventable disease. Major risks factors include tobacco use and alcohol consumption in developed countries and betel quid chewing and bidi smoking in Southeast Asia. The past decade has witnessed significant progress in the understanding of the molecular genetic events underlying the development of HNSCC.4–6 The promise of these studies is the development of novel diagnostic and therapeutic procedures which can be used in the clinical management of these patients.7
HNSCC is a heterogeneous disease with complex molecular abnormalities. In this review we attempt to summarise the molecular mechanisms underlying certain clinicopathological characteristics of squamous neoplasia in the head and neck observed by surgical pathologists. Our goal is to highlight some of the advances made in the understanding of the molecular progression from dysplasia to invasive carcinoma, and the role played by molecular alterations found in non-neoplastic mucosa of patients with HNSCC in local tumour recurrences and the development of second primary HNSCCs. We also survey recent progress in the molecular classification of HNSCC provided by genomic arrays, the molecular biology of HNSCC in “young” individuals, and the relation of human papillomavirus (HPV) and HNSCC.
MOLECULAR PATHOLOGY AND TUMOUR PROGRESSION
Genetic alterations and progression of squamous dysplasia to invasive carcinoma
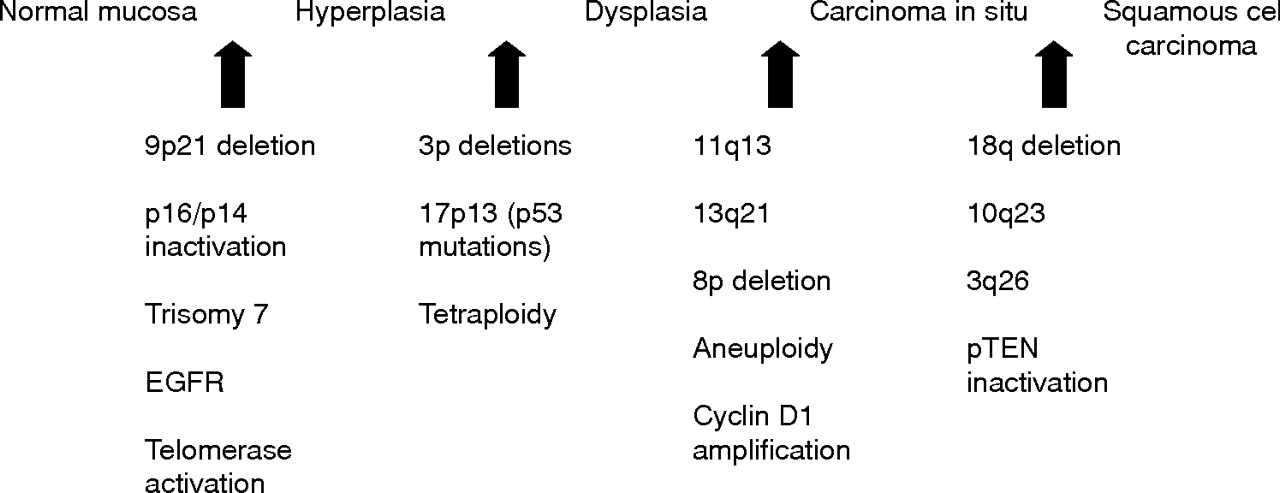
It is generally accepted that HNSCC arises from a common premalignant progenitor followed by outgrowth of clonal populations associated with cumulative genetic alterations and phenotypic progression to invasive malignancy.8–10 These genetic alterations result in inactivation of tumour suppressor genes and activation of proto-oncogenes by deletions, point mutations, promoter methylation, and gene amplification (table 1). Microsatellite marker analysis has allowed the delineation of a genetic progression model for HNSCC based on the frequency of these genetic alterations in preinvasive lesions and invasive tumours (fig 1).9,10 Loss of chromosomal region 9p21 is found in 70–80% of cases, thus representing the most common genetic alteration seen in squamous dysplasia and HNSCC.9,11,12 Loss of heterozygosity (LOH) of 9p21 appears to be an early event in squamous neoplasia of the head and neck and has been found in preneoplastic lesions, including 30% of cases of squamous hyperplasia.11–13 The CDKN2A gene locus found in chromosome 9p21 encodes two different transcripts, p16 and p14ARF, which are responsible for G1 cell cycle regulation and MDM2 mediated degradation of p53. P16 is often inactivated in HNSCC through homozygous deletion, by promoter methylation, and less commonly by point mutations.14
Frequent molecular abnormalities in head and neck squamous cell carcinoma

Hypothetical model for HNSCC carcinogenesis, modified from Califano et al.9
Loss of chromosome region 3p is another common early genetic event in squamous dysplasias and invasive HNSCC.15–17 The specific locus responsible for the tumour suppressor phenotype of 3p remains uncharacterised but investigators have identified at least four distinct regions of allelic loss.15,18 These regions include 3p14, 3p21, 3p22, 3p24, and 3p26. 3p14 contains the fragile histidine triad gene or FIHT, a putative tumour suppressor gene, which has been found to be inactivated by exonic deletions in many tumour types including a small percentage of HNSCC.19,20RSSF1A is another candidate tumour suppressor gene mapped to 3p21 and found to be inactivated by hypermethylation in a small subset of HNSCC.16,21 Much controversy remains regarding the genes present in 3p and their significance and possible roles in HNSCC carcinogenesis. Thus therefore further studies continue.
LOH of 17p and point mutations of the p53 are seen in approximately 50% of HNSCC cases.8,22 Most of these mutations appear to occur late in the progression from epithelial dysplasia to invasive carcinoma.23,24 In a study including 102 invasive carcinomas, 13 severe dysplasias, and 24 cases of squamous carcinoma in situ (CIS), Boyle et al23 reported a prevalence of p53 mutations in 43% of the invasive carcinomas and 19% in the non-invasive lesions. In a longitudinal study of 10 patients with sequential biopsies showing progression from hyperplasia or mild dysplasia to CIS or invasive carcinoma, Shahnavaz et al24 identified p53 mutations in one of 12 dysplasias (8%), two of three CIS cases (66%), and six of eight invasive carcinomas (75%). Nearly 64% of p53 mutations in HNSCC occur in G nucleotides, consistent with exposure to carcinogens such as tobacco.23
Amplification of 11q13 and overexpression of cyclin D1 is seen in 30–60% of HNSCC cases and has been associated with an increased rate of lymph node metastases and overall poor prognosis.25–27Cyclin D1 induces phosphorylation of Rb, thus enabling progression from G1 to S phase. Both amplification of cyclin D1 and inactivation of p16 result in increased phosphorylation of Rb and progression in the cell cycle from G1 to S phase. Amplification and overexpression of cyclin D1 has been described in up to 40% of cases of oral squamous dysplasia, including mild dysplasia.28
In contrast to some colorectal carcinomas, where microsatellite instability is common, in HNSCC this alteration has been identified in only a small subset of preinvasive lesions of the head and neck.29 El-Naggar et al29 reported it in 15% of a small group of head and neck dysplasias. In another study, Ha and coworkers30 showed that it becomes increasingly more common as dysplastic lesions progress to HNSCCs.
Gene expression microarrays suggest that most of transcriptional alterations in head and neck carcinogenesis occur during the transition from normal mucosa to premalignant lesions rather than in the transformation from premalignant lesion to invasive carcinoma.31 When compared with normal mucosa, premalignant lesions showed 108 upregulated and 226 downregulated genes, whereas invasive carcinomas had five upregulated and 13 downregulated genes when compared with premalignant lesions.31
Genetic alterations and risk of progression to malignancy
Leukoplakia and erythroplakia have a well documented risk for transformation into HNSCCs.32,33 The risk of malignant transformation increases with the microscopic finding of dysplasia; however, there is significant lack of agreement in the grading of oral dysplasias.34 One of the promises offered by a better understanding of the molecular basis of preneoplastic lesions of the head and neck is the provision of ancillary tools to provide objective and reproducible grading and estimation of risk of evolving to invasive carcinoma. Oral premalignant tissues frequently show many genomic alterations. In a study by Mao et al,12 51% of patients whose lesions were investigated for LOH at 9p21 and 3p14 loci showed allelic loses at either or both loci. Of the 19 patients with LOH, seven (37%) developed HNSCC while only one of 18 patients without LOH developed HNSCC. In another study of patients with oral leukoplakia, three biomarkers—chromosomal polysomy, suprabasal p53 expression, and LOH at chromosome 3p or 9p—were found to be the most significant predictors of invasive cancer development, in addition to the presence of moderate/severe dysplasia and a past history of oral cancer.35 Rosin and collaborators13 showed differing LOH patterns between non-progressing and progressing low grade oral dysplasias. Both types of lesion were characterised by LOH at 3p or 9p or both; however, patients with progression to in situ or invasive squamous cell carcinomas showed additional losses on 4q, 8p, 11q, or 17p. The same group of investigators have shown that toluidine blue staining identifies premalignant lesions with “high risk” LOH patterns associated with the development of invasive HNSCC.36
Evaluation of DNA ploidy in oral leukoplakia allows the identification of gross genomic alterations and has been show to be a useful tool in identifying lesions with a high risk of malignant transformation.37,38 Sudbø et al37 studied the lesional DNA content in 150 patients with oral leukoplakia and epithelial dysplasia. Thirty six (24%) developed a carcinoma after a mean follow up of 49 months; however, only three of 105 diploid cases (3%) develop invasive disease, as compared with 21 of 25 aneuploid cases (84%) and 12 of 20 tetraploid cases (60%). The negative predictive value for diploid lesions was 97% and the positive predictive value for aneuploid lesions was 84%. DNA content appeared to be an independent prognostic factor, with no statistically significant correlation with the grade of dysplasia. In an earlier study, the same investigators analysed ploidy status of 45 leukoplakic lesions histopathologically negative for dysplasia.38 Thirty five lesions (78%) were diploid, five (11%) were tetraploid, and five (11%) were aneuploid. Four of the five aneuploid cases developed carcinoma (80%) whereas only one of the diploid/tetraploid cases (2%) developed malignancy.38 Patients with aneuploid leukoplakia have a 98% rate of developing primary oral carcinoma, an 81% rate of recurrent or second primary tumours, and a 76% risk of death from disease despite negative histology of the surgical margins.39 Thus ploidy status, chromosome polysomy, and LOH analysis of oral precancerous lesions may provide important information about the risk of progression to cancer, second cancer development, and tumour specific death.
FIELD CANCERISATION, MULTIFOCAL DISEASE, AND SECOND MALIGNANCIES
Field cancerisation and second primary tumours
The concept of “field cancerisation” was first introduced in 1953 by Slaughter et al40 after noting the presence of multiple “independent” tumours and “abnormal epithelium” in the mucosa adjacent to HNSCC. In his now classic study, Slaughter et al40 described multiple carcinomas in 11.2% of 783 unselected cases of HNSCC. Slaughter postulated that “multicentric origin through a process of field cancerisation would seem to be an important factor in the persistence or recurrence of epidermoid carcinoma following therapy”. In its classical interpretation, the concept of “field cancerisation” refers to large areas of the aerodigestive tract mucosa affected by long term exposure to carcinogens, resulting in genetically altered fields in which multifocal carcinomas can develop because of independent genetic events. Many techniques, including chromosome X inactivation, microsatellite analysis, and p53 mutational analysis have confirmed the presence of genetic alterations in the mucosa adjacent to primary HNSCCs, thus providing a genetic explanation for Slaughter’s concept of field cancerisation,9,41–46 but in contrast to Slaughter’s original assertion, these studies strongly suggest that most tumours arising in these abnormal fields are clonally related and originate from a common preneoplastic progenitor. It has been hypothesised that these multiple tumours are caused by the migration of transformed cells through the upper aerodigestive tract mucosa, either by intraepithelial migration or by saliva (micrometastases). The transformed cells have a survival advantage and eventually displace or replace the surrounding mucosa through a process known as “clonal expansion”.9,46
The frequency of genetically abnormal fields in mucosa surrounding HNSCC is difficult to ascertain as most studies addressing this problem have been retrospective, but there is evidence suggesting that it is significantly high. Tabor et al42 found genetically altered fields in the non-neoplastic mucosa of 36% of unselected patients with HNSCC. More importantly, in 70% of these patients, the altered field extended to the surgical margins. Brennan et al47 also detected p53 mutations in 13 of 25 patients (52%) who appeared to have complete tumour resection on the basis of microscopically negative surgical margins; in a different study Tabor et al41 found p53 mutations in 60% of the specimen margins of 10 patients with multiple HNSCC. The size of the abnormal field is difficult to estimate but it can be large and surely varies considerably from patient to patient. Tabor et al41 reported six clonally related tumours separated by 3–6 cm, Worsham et al48 reported two clonally related synchronous carcinomas from the anterior floor of the mouth and pyriform sinus, and Califano et al49 described a patient with multiple squamous cell carcinomas from the hypopharynx and distal oesophagus showing similar allelic loss separated by 40 cm.
Although there is substantial molecular and genetic evidence supporting the concept of field cancerisation, debate remains over the clonal relation of preneoplastic and neoplastic lesions developing in these fields and their mode of spread.6,50,51 Establishing the clonal relation of multiple tumours in the same patient is not always easy or straightforward. Unrelated tumours may lose the same allele by chance alone, and related tumours may show differing allelotypes owing to the accumulation of new alterations during tumour progression. Tabor et al42 demonstrated this conundrum when comparing the genetic alterations between the “field” and the tumours in 28 patients with HNSCC, and found that in only 20% of cases did the tumour and the “field” show similar alterations. In the remaining group, 60% of tumours showed additional losses indicative of genetic progression whereas in 40%, the “field” showed alterations not present in the index tumour.42 In another study, these group of investigators showed discordant genetic alterations between the primary tumour and the corresponding lymph node metastasis in nine of 20 patients (45%). Therefore, similar patterns of LOH with statistical analysis, identical breakpoints, shared microsatellite shifts, and similar p53 mutations have been recommended as evidence of common clonal origin.10,51 Using a combination of current techniques it appears that approximately 60–70% of carcinomas arising in a field originate from a common precursor clone41,46,52,53 whereas the remaining appear to represent “true” multiple primary tumours.45,52,54
Braakhuis et al6,51 have proposed a new classification of secondary HNSCCs arising after treatment of an oral or oropharyngeal carcinoma based on the degree of clonal relation (fig 2). They defined field cancerisation as mucosa with “the presence of one or more areas consisting of epithelial cells that have genetic alterations. A field lesion (or field) has a monoclonal origin, and does not show invasive growth or metastatic behavior”.6 These investigators proposed that tumours with similar genetic profile should be classified as “recurrence” or a metastasis depending on whether the second lesion occurs in the same or a distant anatomical site. If the individual tumours show different genetic alterations, the secondary lesion should be regarded a “true” second primary tumour (SPT). The third group consists of lesions with a common genetic origin but which diverge in later stages; therefore some show similar allelic imbalances and mutations but others show different genotypes. This latter group is classified as “second field tumours” (SFTs).51

Proposed classification by Braakhuis et al51of second tumours after a primary HNSCC.
Molecular pathology of HNSCC and second primary tumours in lung and oesophagus
Approximately 5% of patients with HNSCC develop lung metastases. Given the morphological similarities, the distinction of a primary lung carcinoma from a solitary lung metastasis in a patient with HNSCC cannot be made reliably on histopathological grounds in most cases. To address this problem, Leong et al55 examined the status of chromosomes 3p and 9p in 16 patients with paired resected primary HNSCC and solitary lung carcinomas. Ten of the 16 patients (63%) showed concordant allelic loss at all loci, consistent with a common clonal origin and strongly supporting the conclusion that the pulmonary tumours represented solitary metastases. Three additional samples had different allelic losses consistent with an independent tumour origin, suggesting that the lung lesions were second primary tumours. These results suggest that the majority of solitary lung squamous carcinomas in patients with HNSCC are in fact metastases rather than second primaries, with a smaller number representing second primary tumours. The clonal relation of HNSCC and second “primary” oesophageal squamous cell carcinoma has been investigated by Califano et al49 using microsatellite markers located on chromosomal arms 3p, 9p, and 17p in paired tumours of the head and neck and oesophagus. In 14 cases (83%) the paired tumours had discordant patterns of allelic loss, whereas only two (13%) had identical genetic alterations. Thus most oesophageal carcinomas in patients with HNSSC appear to arise as independent neoplasms with only a small percentage being clonally related.
Detection of minimal disease in surgical margins
Approximately 3.9–32% of patients with HNSCC develop local recurrences despite the presence of microscopically negative margins.56–58 Several studies have investigated the presence and clinical implications of molecular alterations in seemingly tumour-free mucosa in the surgical margins of patients undergoing curative surgery for HNSCC.47,59,60 Detection of genotypically abnormal cells at the surgical margins of resection may be helpful in identifying individuals with a heightened risk of local recurrences after complete surgical resection. In a seminal study addressing this problem, Brennan et al47 demonstrated p53 mutations in 13 of 25 patients (52%) who appeared to have complete tumour resection on the basis of microscopically negative surgical margins. Five of these 13 patients developed local recurrences whereas none of the 12 patients without mutations had recurrences of their tumour. In a study by van Houten et al59 50 of 76 patients (66%) showed p53 mutations in one or more of the surgical margins regarded as “negative” by conventional histopathology. Nine of the patients with p53 mutations developed local recurrences and four developed regional disease. In the group without p53 mutations, there were no local failures and only one patient developed regional disease.59 These investigators found that no other clinicopathological index was predictive of development of local or regional failure. Molecular studies are also helpful in detecting small foci of abnormal mucosa overlooked by conventional histopathological assessment. In a study by van der Toom et al,60 a combination of p53 immunostaining and in situ hybridisation for chromosomes 1 and 7 demonstrated the presence of abnormal cells in 11 of 20 margins (55%) diagnosed as tumour-free in their initial microscopic assessment. After examination of the p53 stains and ISH findings, atypical clusters of 10 to 20 cells that had been diagnosed as normal or hyperplastic became evident. All these cases were upgraded to low grade dysplasia with no case being upgraded to high grade dysplasia or carcinoma.
Data from several studies by Nathan et al61–63 have suggested some prognostic value for overexpression of the proto-oncogene eIF4E in histologically negative margins of HNSCC; eIF4E is a eukaryotic protein synthesis initiation factor increased in almost all HNSCC cases.63 Using immunohistochemistry, Nathan61,62 showed statistically significant differences in local recurrence rates and disease-free intervals between eIF4E positive and eIF4E negative margins; patients with eIF4E-positive margins had a 6.5-fold risk of developing local recurrences. eIF4E was reported to be a more sensitive prognostic indicator for local recurrences than p53 immunohistochemistry.
Gene profiling of head and neck squamous cell carcinoma
The development of high throughput, hybridisation based methods such as cDNA and oligonucleotide microarrays allows the simultaneous analysis of gene expression alterations in thousands of genes in HNSCC.64–71 These studies have used a variety of experimental designs, tumour types, array platforms, and statistical tools, making direct comparisons of results difficult; however, the potential biological and clinical implications of gene expression signatures of HNSCC in tumour classification, prognosis, and treatment are enormous.
The molecular heterogeneity and complexities of subcellular abnormalities in HNSCC have been clearly highlighted by expression studies; however, most of these studies have been small and need independent confirmation in larger groups of patients. In a study of 17 patients with HNSCC, Belbin et al67 identified 375 genes that discriminated between two genotypic subtypes of HNSCC. Patients in group I were younger and had more early stage and paradoxically, poorly differentiated tumours with lower prevalence of nodal metastasis than those in group II; however, 38% of patients in group I died of their cancer whereas none of those in group II died as a result of their cancer after a median follow up period of four years. El-Naggar et al66 analysed 12 matched pairs of normal squamous mucosa and tumour samples from six conventional HNSCC and six variants HNSCC. Parallel expression analysis of 8055 genes showed 26 differentially expressed genes between normal mucosa and tumour specimens. Most of the genes with differential expression were associated with cell differentiation and proliferation. Some of the highly expressed genes in all the tumours included keratin 5, small proline-rich protein 1B (cornifin), S-100 calcium binding protein A8, and keratin 14. Cluster analysis also showed some differences in the overall pattern of expression of certain keratins between conventional HNSCC and variant HNSCC. Conventional HNSCC showed a striking increase in keratins 5 and 14, while none of the variants HNSCC showed changes in those genes. Cornifin was highly expressed in most of the conventional HNSCC but not in the variants. Sarcomatoid and verrucous carcinomas had a pattern of gene expression that was closer to each other than to papillary and basaloid carcinomas. In a different study using a combination of cDNA substraction and microarray analysis, Villaret et al69 found overexpression of 13 genes in HNSCC compared with normal mucosa. These genes included keratin 6, keratin 16, cell adhesion related molecules (plakophilin, laminin, and connexin 26), metalloproteinases (stromelysin-2), p53 regulated elements (14-3-3 sigma protein), and a S-100 calcium binding protein (CaN19). In a study of 60 HNSCCs, Chung et al72 identified four distinct subtypes of HNSCC, based on gene expression. These subtypes included tumours with an EGFR pathway signature, a mesenchymal enriched subtype, a normal epithelium-like subtype, and a subtype with high levels of antioxidant enzymes. The first or EGFR pathway subtype was characterised by overexpression of the genes for transforming growth factor α (TGFα), fibroblast growth factor binding protein (FGF-BP), MMK6, bullous pemphigoid antigen 1, P-cadherin, laminin γ2, and collagen XVII-α, in addition to genes involved in desmosome function (desmoglein 3, desmocollin 2) and keratin 14. The mesenchymal enriched subtype showed high expression of vimentin, syndecan 2, lysyl-oxidase, and four collagen subunits. The normal epithelium-like subtype had the highest expression of microsomal glutathione s-transferase 2 and keratins 15 and 4. The last group included tumours with high expression of glutathione s-transferase M3, thioredoxin reductase 1, and glutathione peroxidase 2 among others. The median follow up period in this study was short, but the investigators reported no significant association between these tumour groups and clinical findings.72
Another line of investigation suggests that gene profiling by cDNA microarrays is capable of identifying upregulated and downregulated genes correlated with tumour recurrences and lymph node metastases (fig 3).73,74 In a study of 20 previously untreated patients, Schmalbach et al73 found significant differential expression of 101 genes between metastatic and non-metastatic oral and pharyngeal carcinomas. The differential profile included genes related to extracellular matrix, cell adhesion and motility, inflammation, and protease inhibition. Collagen type 11 α-1 (COL11A1) and tissue inhibitor of metalloproteinase 1 (TIMP-1) were upregulated in metastatic tumours, whereas serine protease inhibitor-2 (SERPINB2) was downregulated. In a study of 16 oral squamous cell carcinomas, O’Donnell et al75 concluded that metastatic tumours could be differentiated from non-metastatic tumours on the basis of their differential expression of 116 genes. COL2 upregulation was one of the genes associated a “metastatic signature”.75 Ginos et al76 described a “recurrent disease signature” seen in tumours with overexpression of genes implicated in stromal invasion (snail homologue 2, met proto-oncogene), RAS signalling (r-ras homologue), cell surface/extracellular matrix interactions (collagen type IV α5 and α6, laminin γ2, α3, β3; integrin α3, α6, β4, β6) and angiogenesis. All seven patients whose tumours expressed the “recurrent disease signature” failed treatment within 12 months of resection of their primary lesion.76 Using a different technology and data analysis, Warner et al74 found that claudin-1 (CLDN1) overexpression was significantly correlated with lymph node metastases and advanced tumour stage. Roepman and associates77 have recently reported a predictor of lymph node metastases of HNSCC based on the differential expression of 102 genes. Among the predictor genes were genes encoding extracellular matrix components, genes involved in cell adhesion, cell death genes, cell growth and maintenance genes, and genes encoding proteins involved in the degradation of the extracellular matrix. The predictor had an initial overall accuracy of 86% and correctly identified 19 of 22 cases on a subsequent validation set. Interestingly, long term storage of tumour samples affected the predictor performance negatively.

Unsupervised hierarchical cluster analysis of gene expression of 20 oral squamous cell carcinomas on the basis of 1482 genes. Clustering of gene expression is shown on the left, where each row represents one sequence. Genes are shown on the right besides each row. The dendogram shows two groups of tumours. Cluster 1 (left) shows patients with lymph node metastasis compared with cluster 2 (right), which contains patients with early disease stage and no lymph node metastases. Red represents overexpression, green represents underexpression, and black represents no change in gene expression in comparison with a baseline sample (universal RNA, control). (Courtesy of Dr Suzanne Kamel-Reid, Toronto, Canada.)
HUMAN PAPILLOMAVIRUS AND HNSCC
The causal link between human papillomavirus (HPV) and a subset of HNSCC cases has become established in recent years.78–80 HPV DNA has been identified in approximately 15% of HNSCC cases.80–83 The most commonly detected HPV in HNSCC is HPV-16, which has been demonstrated in 90–95% of all HPV positive HNSCC cases, followed by HPV-18, HPV-33, and HPV-33.83–86 The prevalence of HPV DNA in HNSCC varies according to tumour site. The most common location of HPV associated HNSCC (HPV-HNSCC) is the oropharynx. HPV is especially common in carcinomas of the lingual and palatine tonsils, where its reported prevalence has ranged from 45% to 67%.80–83,87,88 A detailed review of HPV infections and tonsillar carcinoma by Syrjänen has been published recently in this journal.89 HPV DNA has been found less often in squamous cell carcinomas of the oral cavity (12–18%), tongue (12%), hypopharynx (13–25%), and larynx (3–7%).82 Epidemiological and case matched studies have also shown that individuals who are seropositive for HPV-16 are at an increased risk for the development of oral or oropharyngeal squamous cell carcinomas.79,86,90 After adjustment for age, sex, and serum cotinine levels, HPV-16 seropositive individuals have a greater risk of development of oral squamous cell carcinomas (odds ratio 2.8) and oropharyngeal squamous cell carcinomas (odds ratio 14.4) than seronegative individuals.79
There is a growing body of evidence suggesting that oropharyngeal HPV-HNSCC is a distinctive clinicopathological and molecular entity.82,91 The risk factors for HPV-HNSCC appear to be different from those of non-HPV HNSCC. Although not universally reported, patients with HPV-HNSCC are less likely to have a history of tobacco use, are more likely to be diagnosed at a younger age, and have a higher frequency of cervical lymph node metastases at presentation. They are also more likely to report a high lifetime number of heterosexual partners, young age at first intercourse, and a history of orogenital sex.85,86,90,91 Most tonsillar HPV-HNSSCs are associated with poorly differentiated, non-keratinising, basaloid morphology, wild-type p53 status, p16INK4A overexpression, and decreased cyclin D1 and Rb expression.80,92–96 High risk oncogenic HPVs such as HPV-16 and HPV-18 encode two oncoproteins, E6 and E7, which induce cellular transformation and dysregulation of cell cycle control. E7 interacts and induces proteolytic degradation of pRb and other RB related “pocket” proteins, whereas E6 inactivates p53 by accelerating its ubiquitin mediated proteolysis.97 In accordance with the oncogenic properties of E6, approximately 90% of HPV-HNSCCs have wild type p53 and lack immunohistochemical overexpression of p53.80,92,98,99 Given the ability of E7 to target Rb for degradation, Rb expression is decreased in HPV-HNSCC (fig 4).92,100 Absence of p16 mutations and overexpression of p16INK4A are also molecular hallmarks of HPV-HNSCC.99–102 It has been suggested that overexpression of p16 may be used as a surrogate marker to identify HPV-HNSCC, and when detected in lymph node metastases is a reliable way to establish an oropharyngeal origin.96,103
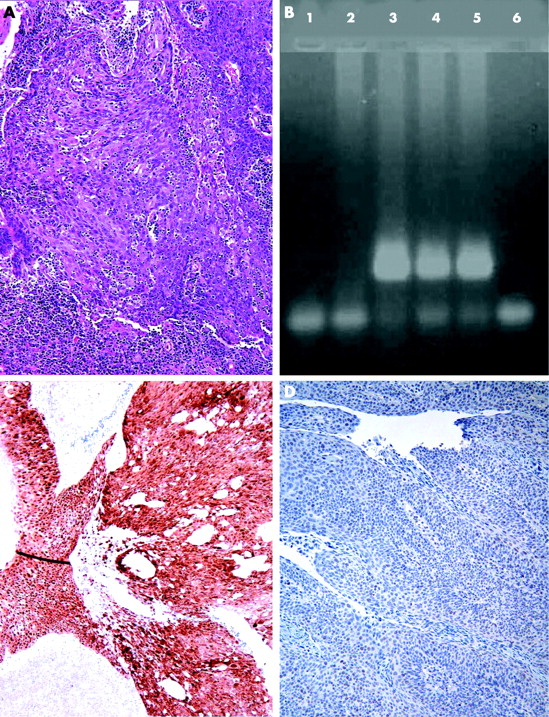
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Poorly differentiated squamous cell carcinoma of tonsil with no evidence of keratinisation (A). Polymerase chain reaction products of DNA extracted from formalin fixed, paraffin embedded tissue show the presence of HPV-16 DNA in lanes 3 to 5 (B). Strong immunoexpression of p16 in same cases (C) with markedly reduced expression of Rb protein (D).
There is strong evidence suggesting that patients with HPV-HNSCC have a more favourable prognosis with lower disease specific mortality than those with HPV-negative HNSCC.80,84,104,105 Gillison et al80 reported a 59% reduction in risk of death from cancer in patients with HPV-HNSCC compared with HPV-negative HNSCC. The detection of HPV in HNSCC has potentially significant therapeutic implications, given the development of vaccines targeting HPV oncogenic proteins E6 and E7 for the prevention of HPV related malignancies.106
MOLECULAR BIOLOGY OF SQUAMOUS CELL CARCINOMA IN YOUNG INDIVIDUALS
Unlike their counterparts in older patients, HNSCC affecting young individuals show an increased incidence in females with no known risk factors, and occur more frequently in the oral cavity and hypopharynx.107–109 The influence of young age on survival has been controversial, with some investigators reporting improved clinical outcome in comparison with older patients,110 whereas most have found no differences on survival.107,109 Such clinicopathological differences suggest that these tumours represent a biologically distinct entity; however, only a few studies have focused on the molecular biology of HNSCC arising in young individuals. In a study comparing “young” and “old” patients, Wang et al108 found a significant greater frequency of microsatellite instability in the young group (88% v 36%). Microsatellite instability was also detected at a higher frequency in tongue (71%) than in laryngeal cancers (23%). The mechanism of microsatellite instability in these tumours was unclear as there were no mutations or promoter methylation of mismatch repair (MMR) genes.
With the possible exception of Fanconi anaemia, the role of HPV in the pathogenesis of HNSCC of young individuals appears to be related to anatomical site of the tumour rather than to age on its own. Sisk and collaborators111 compared the prevalence of HPV between “young” and “old” patients but found no significant differences in the presence of HPV DNA in these two groups. El-Mofty and Lu93 investigated the prevalence of HPV-16 in 33 HNSCCs from young patients. They found a high prevalence of HPV-16 in tonsillar carcinomas (91%). No HPV was found in any of the oral carcinomas and only two of seven laryngeal carcinomas showed HPV DNA.
Fanconi anaemia is an autosomal recessive genomic instability syndrome associated with congenital abnormalities, bone marrow failure, and a predisposition to the development of acute myeloid leukaemia and other solid cancers.112 The relative risk of developing HNSCC in individuals with Fanconi anaemia is 700 times greater than in the general population, the median age at diagnosis of their carcinoma being 26 years.113,114 The risk is greater in patients undergoing haematopoietic stem cell transplantation and in those with acute and chronic graft-versus-host diseases.115 The molecular mechanisms involved in Fanconi anaemia associated head and neck carcinogenesis are not completely understood; however, Kutler et al116 found HPV-16 DNA in a high proportion of HNSCCs affecting individuals with this condition. As is the case with other HPV related HNSCCs, these tumours lacked p53 mutations. These findings support a significant role for HPV in the pathogenesis of HNSCCs arising in the context of Fanconi anaemia.106 The role played by the Fanconi anaemia/BRCA pathway in sporadic HNSCCs, particularly in young individuals with no known risk factors, has not been widely explored; however, in a recent study, Marsit et al100 demonstrated promoter methylation of the FANCF gene in 15% of sporadic HNSCC cases.
CONCLUSION
More traditional molecular studies and genomic arrays will afford us the opportunity for molecular classification of HNSCCs, which will ultimately provide additional insights into HNSCC and possibly new targeted molecular treatments. Despite all these exciting advances, the most effective and cost-efficient tools in reducing the worldwide morbidity and mortality caused by HNSCC are public health policies aimed at reducing or eliminating the use of tobacco products.
Concerns have been recently raised regarding the validity of the results and conclusions regarded in references 37, 38 and 39.