Article Text

Abstract
The complex world of interstitial lung disease presents nearly insurmountable challenges to the general surgical pathologist faced with a lung biopsy in this setting. The pathology is often inflammatory and always requires clinical and radiological context for a relevant and clinically useful histopathological diagnosis. A pattern-based histopathological approach to interstitial lung disease provides a “map” for the general pathologist to navigate this area successfully, especially so when used with aid of the clinical and radiological patterns of presentation.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Statistics from Altmetric.com
Few specimens cause more distress to surgical pathologists than a biopsy sample from a patient with diffuse lung infiltrates. The pathology underlying this clinical and radiological presentation has been referred to as “interstitial” lung disease (ILD) and is nearly always the result of diffuse parenchymal injury, which in turn invokes a stereotypic response of inflammation followed inevitably by repair.1 Unfortunately, there are many ways to injure the lung, and it is the nature of the injury, combined with its acuity, severity and duration, that affects the cellular composition of the tissue response. To complicate matters, any observed histopathology is highly dependent on when the lung biopsy is performed relative to the onset of a given injury. Now, add more than one episode (or type) of injury to the mix and little or no clinical/imaging information, and even the most experienced histopathologist may be ready to send the biopsy specimen out for consultation.
How is the competent and well-trained histopathologist to manage all of these variables when the surgical lung biopsy specimen may appear at most a few times a month in the busiest medical centres? One could read the entire textbook on the subject,2 but I believe that the general pathologist can successfully navigate this complex diagnostic area, without extensive pretraining, by relying on six basic histopathological patterns and an algorithmic approach based on identifying the dominant pattern of disease in the specimen.3 These basic patterns also apply to the much more commonly encountered transbronchial biopsy specimen, but the diagnostic patterns are more limited given the small sample size.4 But, before any lung biopsy is performed, there is a patient with lung disease who is often manageable without a biopsy if one has knowledge of key clinical and radiological patterns of disease.5
THE THREE CLINICAL PATTERNS OF DIFFUSE PARENCHYMAL LUNG DISEASE (ACUTE, SUBACUTE AND CHRONIC)
All successful diagnostic strategies begin with the patient. Before examining the lung biopsy specimen, it is an absolute requirement to know the “tempo” or pace of the patient’s respiratory symptoms. Breathlessness is the main clinical complaint when ILD is present, often accompanied by cough. Knowing whether these symptoms are acute (hours to a several days), subacute (a few weeks to a few months) or chronic (many months to years) allows inclusion of some diseases and exclusion of others from the differential diagnosis. This knowledge also helps us to determine the nature of the critical pathology for this patient (ie, what we should be focusing on in the specimen). Table 1 presents my view of the diseases most commonly associated with these three clinical presentations.
In the patient with acute clinical manifestations, further knowledge about immune status is very helpful, as the index of suspicion for infection is always higher in the immunocompromised host, and the biopsy specimen always requires additional studies to exclude an infectious organism (cultures and special stains for micro-organisms).
THE FOUR RADIOLOGICAL PATTERNS OF DIFFUSE PARENCHYMAL LUNG DISEASE (GROUND GLASS AND CONSOLIDATION, RETICULATION WITH PARENCHYMAL DISTORTION, NODULES AND MOSAIC PATTERNS/CYSTS)
Chest imaging studies (today high-resolution CT (HRCT) is used) figure prominently in the initial clinical evaluation of the patient with ILD because only a limited set of history and physical clues are of independent diagnostic value. Without some knowledge of chest imaging findings, neither clinician nor pathologist has much hope of rendering an accurate and meaningful diagnosis on which to base treatment or additional investigations.
HRCT of the chest also provides invaluable information to the pathologist facing a surgical lung biopsy specimen because pathologists understand gross pathology better than any other specialist in medicine, and the HRCT scan is a reasonable approximation of gross pathology. HRCT provides key information that is useful to the histopathologist with a lung biopsy specimen in hand.5 In the setting of a patient who has undergone a lung biopsy, four basic patterns of radiological lung disease can be discerned: (1) increased attenuation (referred to by our radiology colleagues as “ground glass opacity” and “consolidation”); (2) reticulation with parenchymal distortion (fibrosis); (3) nodules (large or small, singular or multiple); (4) mosaic patterns and cysts. Each of these patterns helps me to interpret the lung biopsy findings (table 2).
THE SIX HISTOPATHOLOGICAL PATTERNS OF ILD (ACUTE INJURY, FIBROSIS, CELLULAR INFILTRATES, AIRSPACE FILLING, NODULES, MINIMAL CHANGES)
In the setting of ILD, it is the rare lung biopsy specimen that has sufficiently unambiguous findings to allow a disease diagnosis (eg, Wegener granulomatosis). In our lung consultation practice, the majority of non-neoplastic lung cases receive descriptive diagnoses, followed by a narrow differential diagnosis and a comment on any additional information that may help to resolve a “clinical–radiological–histopathological” diagnosis. This can be an uncomfortable position for pathologists trained to provide a terse “black or white” diagnosis lest they be accused of “hedging” their risk against error. The reason that naming a specific disease in this setting does not work is that inflammatory processes tend to overlap one another with regard to clinical, radiological, physiological and histopathological features. Despite these stated limitations, the histopathological findings in the lung biopsy do provide critical information about the aetiology, activity, age, reversibility and prognosis of a given case of ILD.
With the microscope, experienced pathologists generally rely on the low magnification pattern of disease, rapidly gaining an overall sense of a histopathological diagnosis using patterns that may not be well articulated by the observer. Unfortunately, ILDs are not biopsied often enough for pathologists to gain first hand experience in view of the exceptionally broad spectrum of this pathology. To circumvent this problem, I teach pathologists to use six basic histopathological patterns in the evaluation of the ILD biopsy specimen. Once the dominant pattern is identified, the differential diagnosis becomes more manageable. Additional findings help to resolve the diagnosis even further. Table 3 presents the six patterns of diffuse lung disease with their respective differential diagnoses.
An algorithmic approach using additional findings can be found online in supplemental table 1. Because of space constraints, I will limit discussion and illustration to the more commonly encountered diseases outlined in these tables. Further exposition of the entities listed in table 3 can be found in a number of excellent textbooks on pulmonary pathology.
We begin with pattern 1 (acute lung injury) because acute clinical symptoms dominate all other concerns regarding the biopsy. Fortunately, these are the easiest cases for pathologists to resolve as long as a few simple rules and a consistent approach is used.
PATTERN 1: ACUTE LUNG INJURY
Basic elements of the pattern: interstitial oedema, intra-alveolar fibrin and reactive type 2 cells (fig 1).

Pattern 1: acute lung injury. At scanning magnification (2× objective), the biopsy sections have an eosinophilic appearance. H&E stain, 15× original magnification.
Key modifiers: hyaline membranes, tissue necrosis, eosinophils and fibrin in alveoli, haemosiderin-laden macrophages, background fibrosis (acute on chronic disease).
Special stains for organisms are required for all lung specimens that show acute injury
Acute lung injury is the histopathological pattern associated with acute clinical lung disease. Onset of symptoms typically occurs hours, days or a week or two before biopsy. Several subtypes of acute lung injury are recognised histopathologically (diffuse alveolar damage (DAD), acute eosinophilic pneumonia, acute fibrinous and organising pneumonia (OP), diffuse alveolar haemorrhage). When specific findings are incomplete (ie, no hyaline membranes), the generic term “acute lung injury” is appropriate.
1a: acute lung injury with hyaline membranes (DAD)
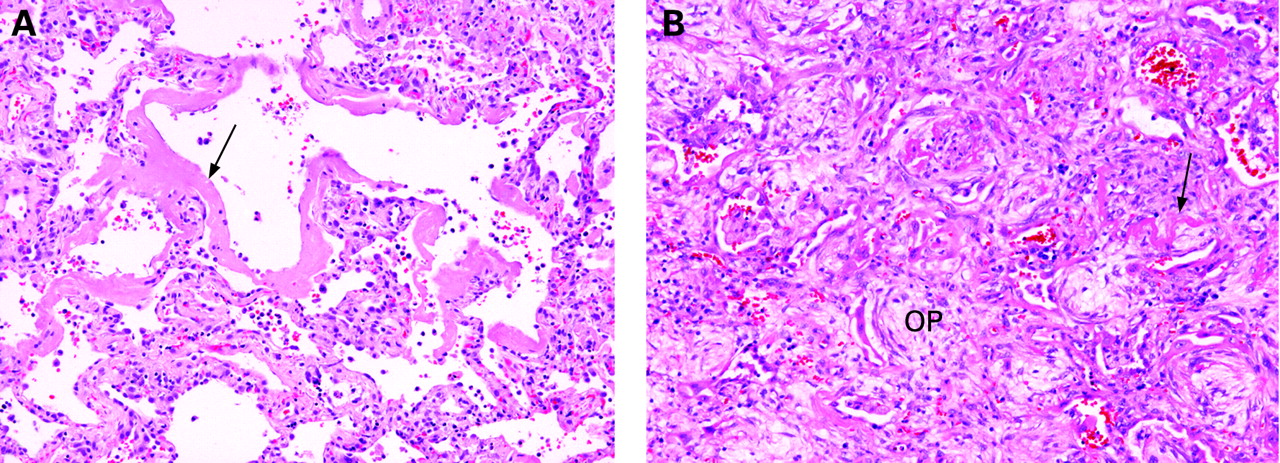
When intra-alveolar hyaline membranes are present, the term “diffuse alveolar damage (DAD)” is appropriate. DAD is a common histopathological pattern of injury in acute diffuse lung disease (fig 2), particularly in patients with clinical adult respiratory distress syndrome and those who are immunosuppressed.7 8 DAD is a diffuse process as the name implies, but it is not always uniform in severity or appearance from one microscopic field to another. DAD is associated with the conditions presented in box 1.

Diffuse alveolar damage. (A) Alveoli are empty and lined by hyaline membranes or (B) variably filled by oedema, macrophages and immature fibroblasts (organisation; OP). Residual hyaline membranes are often present (arrow). H&E stain, 100× original magnification.
Box 1: conditions associated with diffuse alveolar damage78
Infections (viral, fungal, bacterial, parasitic)
Toxic inhalants
Drug reactions
Shock
Systemic collagen vascular diseases
Radiation reactions (acute)
Acute allergic reactions (eg, hypersensitivity pneumonitis)
Alveolar haemorrhage syndromes
Miscellaneous conditions
Idiopathic disease (acute interstitial pneumonia)
In the immunocompromised patient, infection leads the differential diagnosis. DAD can also occur in patients with idiopathic pulmonary fibrosis and other chronic ILDs, possibly as a natural component of the disease evolution.9 10
1b: acute lung injury with necrosis
Tissue necrosis raises a differential diagnosis of infection, infarction, neoplasm and Wegener granulomatosis (and similar conditions such as so-called necrotising sarcoidosis and sometimes Churg–Strauss syndrome). When necrosis is present (fig 3) in the acutely ill patient, infection leads the differential diagnosis, even if special stains are negative. Conversely the lack of necrosis in a biopsy specimen from an acutely ill patient with normal immunity virtually eliminates clinically significant infection as the primary aetiology in my experience.

Acute injury with necrosis. Necrosis (N) is a harbinger of infection in the context of pattern 1 and an acutely ill patient. Infection always leads the differential diagnosis in this situation, even if special stains are negative. H&E stain, 100× original magnification.
1c: acute lung injury with alveolar eosinophils
Tissue eosinophils play important roles in a number of toxic, infectious and immunological lung reactions.11 When many eosinophils are visible in the airspaces (fig 4) in a patient with acute lung disease, a diagnosis of acute eosinophilic pneumonia is appropriate.12 This finding is a vital key to potentially reversible disease and should never be discounted or overlooked. Potential causes of eosinophilic pneumonia are presented in box 2.

Acute eosinophilic pneumonia. (A) When many eosinophils are visible in the airspaces in a patient with acute lung disease, a diagnosis of acute eosinophilic pneumonia is appropriate. (B) Organisation in alveolar spaces, and rarely hyaline membranes, may be present. (A,B) H&E stain, 100× original magnification.
Box 2: causes of eosinophilic lung disease13
Certain infections and parasitic diseases
Asthma and asthma-related lung diseases
Churg–Strauss syndrome
Drug reaction
Acute eosinophilic pneumonia (idiopathic)
Acute smoking-related disease (rare)
1d: acute lung injury with diffuse alveolar haemorrhage
Evidence of recent and remote haemorrhage in the lung is the most important histopathological feature for distinguishing immunologically mediated haemorrhage syndromes from other forms of acute lung injury.14 Diseases such as Wegener granulomatosis and Goodpasture syndrome may show considerable histopathological overlap with those producing DAD.15 In most instances, pulmonary haemorrhage is recognised as the clinical problem because the patient has experienced one or more episodes of haemoptysis, but this can be an inconsistent finding.
At scanning magnification, the biopsy specimen of the diffuse alveolar haemorrhage lung has variable fresh blood in the parenchyma, typically associated with fibrin and haemosiderin-filled alveolar macrophages (fig 5). In more chronic examples, this dense macrophage reaction may even mimic the idiopathic interstitial pneumonia referred to as “desquamative interstitial pneumonia” (DIP) (see below). OP is common in this setting and is often associated with fibrin in the alveoli, the latter being vital for the correct assessment that one is dealing with an acute process (alveolar fibrin is not an expected finding in idiopathic DIP). The most common pulmonary haemorrhage syndromes and other disease processes associated with diffuse pulmonary haemorrhage are presented in box 3.

Diffuse alveolar haemorrhage. (A) The lung biopsy specimen has variable fresh blood in the parenchyma, typically associated with fibrin, reactive type 2 cells and haemosiderin-filled alveolar macrophages (B). This reaction pattern may be associated with immunologically mediated injury. (A,B) H&E stain; (A) 40× original magnification; (B) 400× original magnification.
Box 3: causes of diffuse alveolar haemorrhage14
Goodpasture syndrome (anti-glomerular basement membrane antibody disease)
Vasculitides (especially Wegener granulomatosis)
Mitral stenosis
IgA nephropathy
Behcet syndrome
Certain systemic collagen vascular diseases (especially systemic lupus erythematosus)
HIV infection
Anti-phospholipid syndrome
Pulmonary veno-occlusive disease
Idiopathic pulmonary haemosiderosis
Drug reactions, including toxic reactions and anticoagulants
Acute lung allograft rejection
Unclassified forms
Capillaritis is a distinctive histopathological feature seen in some alveolar haemorrhage syndromes (fig 6). Capillaritis can be quite focal in the biopsy specimen and is especially important to identify once the other features of alveolar haemorrhage are encountered because the presence of capillaritis is considered a medical emergency and requires an immediate call to the clinician (patients are at risk of fatal haemoptysis).

Capillaritis in diffuse alveolar haemorrhage. (A) The presence of capillaritis is one of the few medical emergencies in pulmonary pathology. Capillaritis is distinctive but can be quite focal in the biopsy specimen. Here a “pseudo-bronchopneumonia” pattern is present, with shed neutrophils filling alveolar spaces. (B) Here the neutrophils are still within the alveolar walls. (A,B) H&E stain; (A) 40× original magnification; (B) 400× original magnification.
Serological studies are essential in documenting an immunological mechanism and providing information on the presence of a specific haemorrhage syndrome (eg, anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies, anti-glomerular basement membrane antibodies). We do not perform tissue immunofluorescence studies routinely in biopsy material, mainly because the serological data are more consistent, quite reliable, and preferred for treatment selection by many clinical experts today. Sometimes the pigmented alveolar macrophages of smokers can simulate the siderophages of pulmonary haemorrhage. The fine granularity of the brown pigment in these cells and the consistent presence of dot-like black pigment particles in the cytoplasm helps in their proper identification.
1e: acute lung injury with background fibrosis (acute on chronic disease)
Acute lung injury can occur as a natural escalation of an underlying chronic lung disease, such as acute exacerbation of idiopathic pulmonary fibrosis.9 10 Alternatively, a patient may have a stable chronic lung disease on which community-acquired pneumonia or drug toxicity is now superimposed. I refer to this combination of histopathology as “acute on chronic lung disease” and always append a comment regarding the diagnostic possibilities in this scenario (one disease versus two or more).
PATTERN 2: FIBROSIS
Basic elements of the pattern: dense collagen deposition in the lung parenchyma, often accompanied by some degree of structural remodelling with alveolar loss (fig 7).

Pattern 2 fibrosis. Fibrosis in the lung parenchyma should be diagnosed only when it is dense, and never in the transbronchial biopsy specimen. Note the distortion of the alveolar parenchyma and fusion of alveolar walls.
Key modifiers: with temporal heterogeneity, with uniform alveolar wall fibrosis, with airway-centred scarring, with isolated stellate scars, with microscopic honeycombing only, with pleuritis.
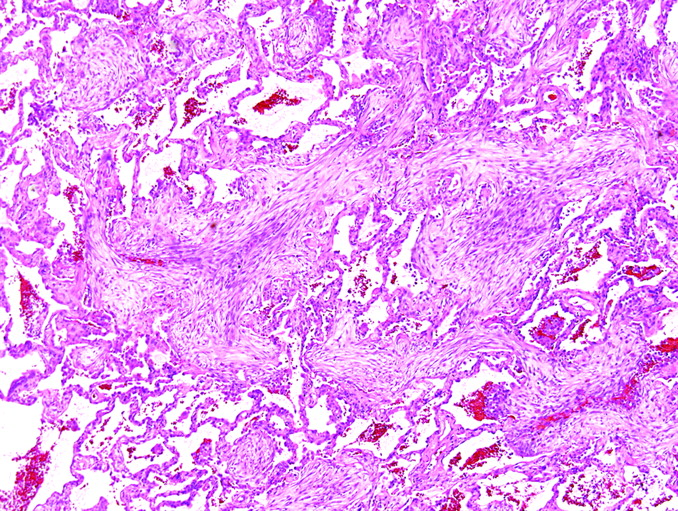
Interstitial lung fibrosis is often accompanied by permanent and irreversible alteration of lung architecture. Pattern 2 (fibrosis) tends to carry great prognostic significance for the patient and is only superseded in importance by pattern 1 (acute lung injury). Different patterns of fibrosis probably derive from different injury mechanisms, carry different prognostic implications, and one day may influence targeted treatments. Large, often stacked, cystic spaces that can be seen on CT scans of the chest and in whole-lung sections and referred to as “honeycomb cysts” are often recapitulated (or preceded) at the microscopic level, where the process is referred to as “microscopic honeycombing” (fig 8). A general morphological approach to diffuse lung fibrosis should include an assessment of the distribution and character of the fibrotic or fibroblastic reaction, the degree and extent of mature interstitial scarring, and the presence or absence of microscopic honeycomb remodelling.

Honeycomb remodelling. (A) Large, often stacked, cystic spaces seen in whole-lung sections are referred to as “honeycomb cysts”. (B) These are often recapitulated (or preceded) at the microscopic level, where the process is referred to as “microscopic honeycombing”. (A) Gross image; (B) H&E stain, 1× original magnification.
2a: fibrosis with temporal heterogeneity
Usual interstitial pneumonia (UIP) is the prototypic chronic interstitial pneumonia with “temporally heterogeneous” interstitial fibrosis and honeycombing (both microscopic and macroscopic), originally described by Liebow and Carrington.16 Patients with cryptogenic fibrosing alveolitis have UIP on surgical lung biopsy.17 UIP is characterised by zones of normal lung tissue adjacent to zones of advanced architectural remodelling (fig 9).18 The latter is recognised by confluent and dense scarring of the alveolar parenchyma. Microscopic honeycombing occurs early in the process and consists of irregular cysts containing mucus, aggregated in dense fibrosis (fig 10). For me, microscopic honeycombing requires fibrosis on at least three sides of the aggregated cysts, a criterion that helps to avoid including foci of peribronchiolar metaplasia under this designation. Small discrete foci of active fibroplasia are always present in UIP, but they are not specific for UIP. These “fibroblastic foci” occur at the interface between dense scar and adjacent normal lung (fig 11). These three key elements of UIP are often related to one another in the biopsy specimen, as a transition from old disease (fibrosis) to normal lung occurs, with active “fibroblast foci” forming a leading edge between them (this is the concept underlying the term “temporal heterogeneity”—heterogeneous in time—with yesterday’s lung destroyed by fibrosis, tomorrow’s lung waiting to be consumed, and fibroblast foci sitting at the interface (today)). A peripheral acinar pattern can often be recognised in UIP, accompanied by relative centriacinar sparing. These findings help to distinguish UIP from other lesions (see below) with interstitial fibrosis and honeycombing (box 4). Rarely, other diseases can simulate the “UIP pattern”, such as chronic hypersensitivity pneumonitis, the rheumatic diseases and asbestosis.

Usual interstitial pneumonia (UIP). UIP is characterised by zones of normal lung tissue adjacent to zones of advanced architectural remodelling (temporal heterogeneity). (A) Early in the disease process, an interrupted “rind” of subpleural fibrosis is visible. (B) A more advanced stage shows more extensive perilobular fibrosis with relative centrilobular sparing, producing ring-like scarring at scanning magnification. (A,B) H&E stain, 1× original magnification.

Microscopic honeycombing. This finding occurs early in usual interstitial pneumonia and consists of irregular microscopic cysts containing mucus (with neutrophils) and lined by ciliated columnar epithelium. H&E stain, 40× original magnification.

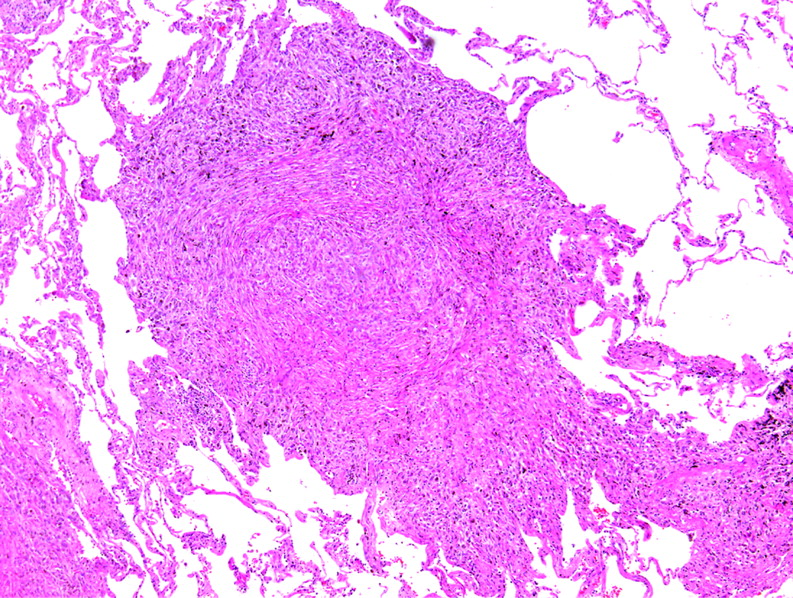
Fibroblast foci. Discrete “bulges” of immature fibroblasts in amphophilic matrix are referred to as “fibroblast” or “fibroblastic” foci. They occur at the interface between dense scar and adjacent normal lung. In a three-dimensional reconstruction study, the foci appear to be interconnected into a lattice. These lesions are thought to be the focus of ongoing injury and repair in usual interstitial pneumonia (UIP), but are not unique to UIP. H&E stain, 200× original magnification.
Box 4: diseases with fibrosis/honeycombing (modified from Leslie et al1)
Idiopathic pulmonary fibrosis (idiopathic usual interstitial pneumonia)
Desquamative interstitial pneumonia
Lymphoid interstitial pneumonia
Systemic collagen vascular disease
Chronic drug reactions
Pneumoconioses (asbestosis, berylliosis, silicosis, hard metal pneumoconiosis, others)
Sarcoidosis
Pulmonary Langerhans cell histiocytosis (histiocytosis X)
Chronic granulomatous infections
Chronic aspiration
Chronic hypersensitivity pneumonitis
Organised, and organising, diffuse alveolar damage
Chronic interstitial pulmonary oedema/passive congestion
Radiation injury (chronic)
Healed infectious pneumonias and other inflammatory processes
Non-specific interstitial pneumonia/fibrosis
Erdheim–Chester disease
Hermansky–Pudlak syndromes
2b: fibrosis with uniform alveolar wall involvement
The occurrence of “interstitial” fibrosis that tends to preserve alveolar structure (ie, little confluence of scar) characterises a fairly limited group of ILDs, dominated by rheumatic diseases, chronic drug reactions and some examples of chronic hypersensitivity (fig 12). An idiopathic form (referred to as “fibrotic non-specific pneumonia” or simply “NSIP”) was formally described by Katzenstein and Fiorelli19 in 1994, who reported on 64 patients whose lung biopsy specimens showed cellular interstitial inflammatory changes that did not fit within the spectrum of diseases originally described in the Liebow historical classification of the idiopathic interstitial pneumonias. In their report, they coined the term “non-specific interstitial/fibrosis (NSIP/F)” for the patterns identified, and openly recognised that these patterns probably represented a wide variety of inflammatory processes affecting the lung. These authors emphasised the temporally uniform appearance of the disease process—that is, the pathology seemed to reflect a single injury in time (ie, lacking a spectrum ranging from new disease to old). Perhaps the most important aspect of the Katzenstein and Fiorelli study was the discovery that, in patients with NSIP, morbidity and mortality were significantly different from that expected for UIP.20

Diffuse alveolar wall fibrosis. Sometimes “interstitial” fibrosis preserves alveolar structure (ie, little confluence of scar) invoking a fairly limited differential diagnosis, dominated by rheumatic diseases, chronic drug reactions and some examples of chronic hypersensitivity. When no underlying aetiology is present, the term “idiopathic NSIP” is appropriate. H&E stain, 40× original magnification.
2c: fibrosis with an airway-centred distribution
When scarring occurs diffusely around bronchioles (fig 13), the differential diagnosis generally is limited to inhalation (eg, hypersensitivity pneumonitis)21 and aspiration-associated injury and certain rheumatic or immune-mediated systemic conditions (eg, rheumatoid arthritis, Sjogren syndrome). An idiopathic form has been described.22 23 In some biopsy samples, the airway-centred nature of the process may be difficult to discern, especially when fibrosis is advanced and/or the sample is small. The HRCT distribution may be helpful, as UIP and the autoimmune diseases tend to involve the periphery and lower lung zones, whereas diffuse inhalational injuries tend to have a more mid-zone and upper lung zone distribution (at least relatively early in the process).

Airway-centred fibrosis. Fibrosis around bronchioles is typically a manifestation of inhalational or aspiration injury to the lungs. H&E stain, 15× original magnification.
2d: fibrosis with isolated stellate scars
The late stages of the smoking-related lung disease known as Langerhans cell histiocytosis (LCH) are characterised by the presence of stellate parenchymal scars (fig 14).24 These scars are distinctive and typically have few or no residual Langerhans cells. We refer to these as “healed” lesions of LCH. They may be incidental when the biopsy is performed for localised disease (such as carcinoma). In the setting of ILD, a form of “smoking-related interstitial lung disease” should be considered as the correct diagnosis.

Langerhans cell histiocytosis (LCH). The star-shaped, airway-centred scars of LCH are distinctive and may be a sign of resolved disease. H&E stain, 15× original magnification.
2e: fibrosis with microscopic honeycombing only
Many unrelated lung diseases can result in localised areas of complete structural remodelling (end-stage lung) with the formation of microscopic honeycomb cysts in fibrosis. Context is essential. If microscopic honeycombing dominates the entire biopsy sample, the patient is over 60 years of age, and peripheral bibasilar fibrosis is present on HRCT, the correct diagnosis is nearly always cryptogenic fibrosing alveolitis. Nevertheless, in this setting, pathologists should use the descriptive term “advanced microscopic honeycomb remodelling only”, because the UIP diagnosis today requires some normal preserved lung in the biopsy specimen to establish “temporal heterogeneity”.
2f: fibrosis with pleuritis
The pleura is an organ separate from the lung. When the lung biopsy sample shows fibrosis, and the pleura is actively inflamed (acute or chronic), always consider one of the rheumatic diseases as a potential aetiology.
PATTERN 3: CHRONIC INFLAMMATORY (CELLULAR) INFILTRATES
Basic elements of the pattern: chronic inflammatory cells present diffusely within alveolar walls, often with variable intensity (fig 15).

Pattern 3: chronic inflammatory (cellular) infiltrates. The biopsy specimen in pattern 3 tends to have a blue appearance because of the interstitial infiltrates of mononuclear cells (mainly lymphocytes and plasma cells) with their haematoxylin-stained nuclei and scant cytoplasm. H&E stain, 15× original magnification.
Key modifiers: with pure cellular interstitial pneumonia, with poorly formed granulomas, with well-formed granulomas, with diffuse alveolar wall fibrosis (see pattern 2b), with confluent dense fibrosis (see pattern 2a; simulators of the UIP pattern)
Diffuse infiltration of the lung parenchyma by inflammatory cells (typically including lymphocytes and plasma cells) is a very common pattern among ILDs, and often not particularly helpful in arriving at a specific diagnosis. The diseases that produce mononuclear interstitial infiltrates tend to overlap in their response to treatment and patient prognosis. The more common diseases in this pattern are presented here.
3a: cellular infiltrates with pure cellular interstitial pneumonia (lymphocytes and plasma cells)
Cellular interstitial pneumonia patterns of ILD had been recognised by pulmonary pathologists for many years, but they lacked a champion until 1994, when Katzenstein and Fiorelli described their 64 patients with NSIP.19 Three histopathological patterns were evident in their series. One was a pure cellular form (group 1; fig 16), whereas two others had variable interstitial fibrosis (discussed under pattern 2b). In practice, this “group 1” of NSIP is rare.

Non-specific interstitial pneumonia (NSIP), cellular. The pure cellular form of NSIP is rare. If small poorly formed granulomas are present, hypersensitivity pneumonitis should be considered. If foci of organising pneumonia are present, without granulomas, cryptogenic organising pneumonia should be considered. H&E stain, 40× original magnification.
3b: cellular interstitial pneumonia with poorly formed granulomas
The prototype of this pattern of ILD is hypersensitivity pneumonitis (HP) to inhaled organic antigen, also known as extrinsic allergic alveolitis (fig 17).25–28 Other inflammatory lung diseases can mimic HP in both the subacute and chronic forms of the disease. I always include drug reaction, systemic autoimmune disease and even evolving low-grade lymphoproliferative disease in the differential when this pattern is encountered. Helpful HRCT findings favouring subacute HP (the form most consistently cellular in lung biopsy specimens) include the presence of ill-defined centrilobular nodules in the mid and upper lung zones.29 The idiopathic interstitial pneumonia corresponding to this pattern is referred to as “lymphoid interstitial pneumonia (LIP)”. Lymphoma must be excluded before a diagnosis of LIP can be rendered.

Hypersensitivity pneumonitis (HP). The cellular interstitium of subacute HP (A) is dominated by plasma cells (magnification in B). A typical poorly formed granuloma of HP is present in (B). (A,B) H&E stain; (A) 40× original magnification; (B) 400× original magnification.
3c: cellular interstitial pneumonia with well-formed granulomas
Infection dominates this pattern of cellular interstitial pneumonia, followed by subacute/chronic aspiration pneumonia, especially if the granulomas are present in alveoli or alveolar ducts (fig 18). Atypical mycobacteria dominate this diffuse lung disease presentation, including a form of bioaerosol exposure to atypical mycobacteria (so-called “hot tub lung”).30 When granulomas are interstitial and resemble those of sarcoidosis (see below), the presence of cellular infiltrates is best reconciled as a different disease process (eg, drug reaction and sarcoidosis).

Granulomatous pneumonia from infection. This example of atypical mycobacterial infection (Mycobacterium avium complex) differs from hypersensitivity pneumonitis in having larger and better formed granulomas, along with more granulomas in the alveolar spaces and alveolar ducts. Necrosis in granulomas may be present (not in this image) and is a harbinger of infection. Sarcoidosis granulomas (fig 25) are better formed, have less associate inflammation, and consistently have more hyaline fibrosis around aggregated granulomas. H&E stain, 40× original magnification.
PATTERN 4: ALVEOLAR FILLING
Basic elements of the pattern: alveoli in the biopsy specimen filled with cells or non-cellular material (fig 19).

Pattern 4: alveolar filling. This example of diffuse alveolar haemorrhage nicely demonstrates the concept of alveolar filling. In cases of inflammatory reaction to injury, initial interstitial changes typically evolve to alveolar filling disease (eg, acute bronchopneumonia, organising pneumonia). H&E stain, 15× original magnification.
Key modifiers: with immature fibroblasts, with macrophages, with proteinaceous material, with blood and siderophages, with neutrophils.
This pattern of lung disease occurs as a component of a number of different pathological processes such as acute and organising infections, pulmonary haemorrhage, pulmonary alveolar proteinosis (PAP), chronic eosinophilic pneumonia, DIP, respiratory bronchiolitis-associated interstitial lung disease (RB-ILD) and many others. Parenchymal consolidation alone is not helpful in the differential diagnosis except when the filling process is distinctive or nearly diagnostic, such as PAP (granular proteinaceous material) or chronic eosinophilic pneumonia (pink macrophages, fibrin and eosinophils).
4a: alveolar filling with immature fibroblasts (OP pattern)
The OP pattern is a very common reaction pattern in the lung. The presence of intraluminal tufts of plump fibroblasts and immature connective tissue within alveolar ducts and more distal airspaces has been traditionally referred to as “bronchiolitis obliterans organising pneumonia” or “BOOP” by pathologists. Today we use the term “OP pattern” as a more generic descriptor of the lesion and to avoid potential confusion with “idiopathic BOOP” (now cryptogenic organising pneumonia (COP)). An OP pattern is especially evident in organising acute lung injury from any aetiology. OP can be accompanied by alveolar fibrin and/or hyaline membranes if acute injury is ongoing. This morphology typically will be associated with acute illness, whereas the patient with a pure OP pattern will often have a more subacute presentation.
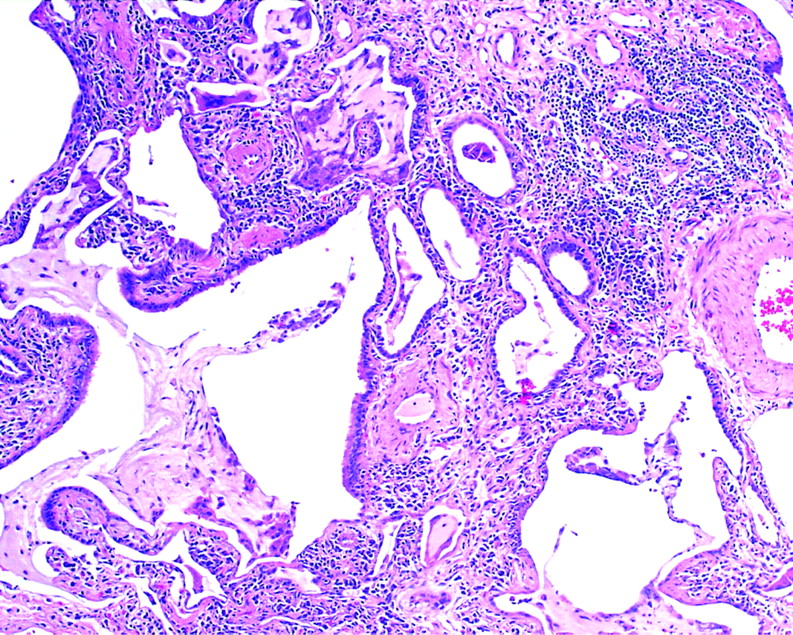
OP is seen in a number of settings (see below), most notably in COP, a form of idiopathic ILD.31 The most consistent finding in COP is patchy involvement of the airspaces by small tufts of immature fibroblasts distributed within terminal bronchioles, alveolar ducts and alveoli (fig 20). Other findings that may accompany an OP pattern include interstitial infiltrates of mononuclear cells, fibrinous exudates, foam cells in the airspaces and prominent type II pneumocytes. Common causes of an OP pattern are presented in box 5.
Cryptogenic organising pneumonia (COP). The most consistent finding in COP is patchy involvement of the airspaces by small tufts of immature fibroblasts distributed within terminal bronchioles, alveolar ducts and alveoli. H&E stain, 40× original magnification.
Box 5: common causes of the organising pneumonia pattern (modified from Leslie et al)1
Organising infections (any cause)
Organising diffuse alveolar damage
Hypersensitivity pneumonitis
Organising infectious pneumonias in:
chronic bronchitis and emphysema
bronchiectasis
cystic fibrosis
aspiration pneumonia
chronic bronchiolitis
Drug and toxin reactions
Systemic collagen vascular diseases
Eosinophilic pneumonia
Airway obstruction
Cryptogenic organising pneumonia
Peripheral reaction around:
abscesses
infarcts
Wegener granulomatosis
others
4b: alveolar filling with macrophages (DIP-like reaction)
A DIP-like pattern is characterised by increased numbers of alveolar macrophages, with mild associated inflammatory changes in alveolar walls (fig 21). Lesions that may show a DIP pattern (in some cases focally) are presented in box 6.

Desquamative interstitial pneumonia (DIP). Idiopathic DIP is characterised by dense alveolar macrophages. Many conditions can result in localised alveolar macrophage accumulation. Inset: alveolar macrophage detail. H&E stain, 40× original magnification (inset, 400× original magnification).
Box 6: conditions associated with a desquamative interstitial pneumonia (DIP)-like reaction (modified from Leslie et al1)
Obstructive pneumonias (with foamy alveolar macrophages)
Exogenous lipoid pneumonia and lipid storage diseases
Infection in the immunosuppressed patient (“histiocytic pneumonia”)
Respiratory bronchiolitis-associated interstitial lung disease
Pulmonary Langerhans cell histiocytosis
Drug reactions
Chronic alveolar haemorrhage
Eosinophilic pneumonia
Certain pneumoconioses (especially talcosis, hard metal disease and asbestosis)
Idiopathic DIP
The idiopathic form of DIP described by Carrington represents a distinct pathological entity that has clinical, radiological and prognostic differences from idiopathic UIP.32 Some cases previously classified as DIP can be reasonably reclassified as RB-ILD, an ILD of smokers that does not appear to progress to advanced fibrosis.33
The cytological features of the macrophages in all of these conditions vary considerably and are helpful at times in pointing to a specific diagnosis. In RB-ILD, the macrophages are airway-centred and contain fine, light-brown, cytoplasmic pigmentation with delicate black punctation, findings characteristic of smokers’ macrophages. In amiodarone reactions, obstructive pneumonias, lipoid pneumonia and storage diseases, foamy or vacuolated histiocytes predominate. In hard metal disease (cobalt pneumoconiosis), distinctive multinucleated intra-alveolar histiocytes are the dominant finding.34 Chronic alveolar haemorrhage is associated with extensive haemosiderin-laden macrophages in the airspaces.14 The distinctive features of eosinophilic pneumonia are the presence of interstitial and airspace eosinophils, airspace fibrin, markedly reactive type II cells and dense alveolar macrophages.12 Birefringent material can be identified within the DIP-like reaction in many of the pneumoconioses. Large and small clear spaces, often engulfed by giant cells and associated with variable fibrosis, characterise exogenous lipoid pneumonia (fig 22).
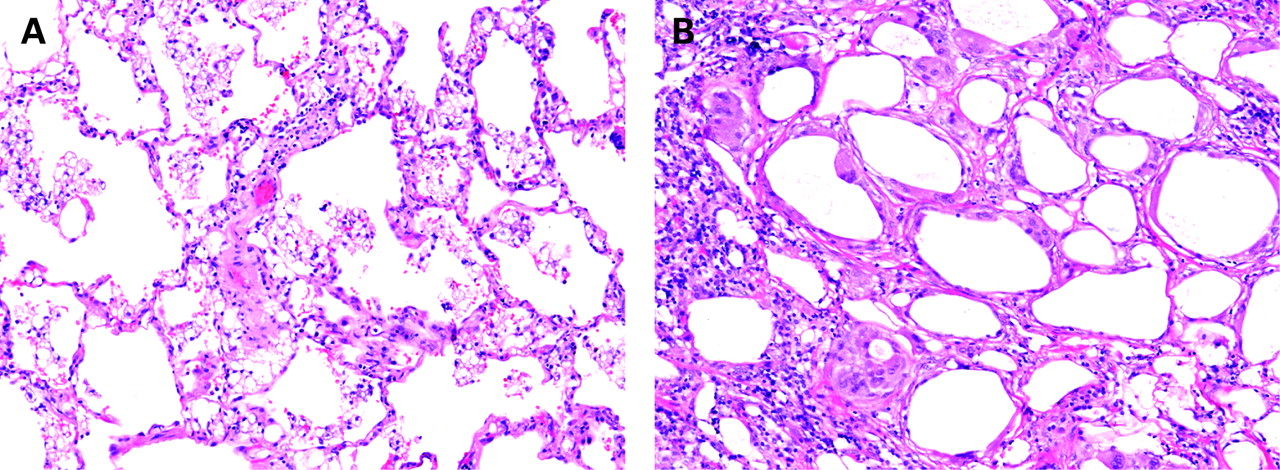
Exogenous lipoid pneumonia. The microscopic appearance of exogenous lipoid pneumonia is dependent on the composition of the aspirated material. (A) Exogenous lipoid pneumonia with histiocytes containing predominantly fine microvesicles. (B) Exogenous lipoid pneumonia with larger vacuoles and associated fibrosis. (A,B) H&E stain, 40× original magnification.
4c: alveolar filling with eosinophilic material
The prototype of this pattern is pulmonary alveolar proteinosis (PAP). This distinctive disease results in dense granular eosinophilic material filling adjacent alveoli. PAP can be focal or segmental in distribution. Cholesterol clefts and hyaline globules are typically present in the granular infiltrates, and a rim of retraction often separates the infiltrates from adjacent alveolar walls (fig 23). The disease occurs commonly as a primary idiopathic form, but may also be seen as a secondary phenomenon in the settings of occupational disease (especially dust-related), drug-induced injury, haematological diseases and in many settings of immunodeficiency.35 36 The granularity of the alveolar material helps differentiate PAP from other alveolar filling processes (such as pulmonary oedema and Pneumocystis infection).

Pulmonary alveolar proteinosis (PAP). All of the diagnostic features of PAP are identifiable in this image: alveolar exudates with an eosinophilic granular appearance, scattered larger inclusions with more intense eosin staining, and slight retraction effect at the periphery of the alveolus. H&E stain, 40× original magnification.
4d: alveolar filling with blood and siderophages
Alveolar haemorrhage syndromes (discussed under pattern 1d) are the diagnosis of exclusion for this alveolar filling pattern. Sometimes striking alveolar haemorrhage can be seen in urgent lobectomy specimens from patients with persistent haemoptysis related to bronchiectasis or other airway/vascular abnormality. As always, the clinical context is essential, as this is most often a localised (ie, lobar) phenomenon.
4e: alveolar filling with neutrophils
Acute infectious bronchopneumonia is the prototype for alveolar filling with neutrophils. This pattern is most commonly identified in autopsy material and rare in surgical biopsy specimens. When this pattern is seen very focally in the specimen, consider other causes of neutrophil exudation, particularly capillaritis in diffuse alveolar haemorrhage.
PATTERN 5: NODULES
Basic elements of the pattern: nodules in the biopsy specimen, well or poorly formed, large or small, single or numerous (fig 24)
Pattern 5: nodules. In pattern 5, nodules may be well or poorly formed, large or small, single or numerous. This is a patient with sarcoidosis. H&E stain, 15× original magnification.
Key modifiers: with lymphoid cells, with atypical cells, with well-formed granulomas, with macrophages and dust, with Langerhans cells and stellate scars, with OP.
Pattern 5 is dominated by neoplastic diseases, especially when a single or limited number of unilateral nodules are present on HRCT. The spectrum of isolated neoplastic disease is beyond the scope of this work and is not discussed. When nodules are small, bilateral and numerous, nodular forms of ILD enter the differential diagnosis. Randomly distributed nodular lesions often dominate the pathology of miliary infections such as miliary tuberculosis or other disseminated infections. We will explore three types of diffuse nodular lung disease: granulomas, nodules in silicate disease and nodular LCH.
5a: nodules with granulomatous inflammation
Relatively few diffuse interstitial pneumonias are dominated by the presence of granulomas, either necrotising or non-necrotising in type. These are presented in box 7.
Box 7: diffuse diseases associated with nodular granulomatous inflammation37
Granulomatous infections
Sarcoidosis
Rheumatoid nodules
Intravenous talcosis
Pneumoconioses (eg, inhalation talcosis, berylliosis)
Aspiration pneumonia
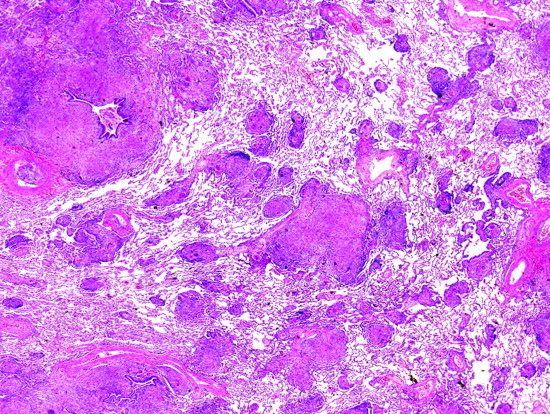
Specific clues to the aetiology of granulomatous interstitial pneumonias include the anatomical distribution, and the qualitative features of the granulomas themselves.38 In sarcoidosis and berylliosis, conglomerates of non-necrotising granulomas are present in a distribution following lymphatic routes.39 These granulomas tend to be surrounded by dense, brightly eosinophilic lamellar collagen, and adjacent granulomas have a tendency to coalesce within this matrix (fig 25). Infectious granulomas may be solitary or confluent, and may or may not be associated with necrosis. Necrosis, particularly if microabscess-like, should raise suspicion for infection and lead to rigorous exclusion with special stains and cultures. Although necrosis may be seen rarely in the granulomas of sarcoidosis and berylliosis, as a rule, the necrosis in the latter granulomas has a more fibrinoid or hyaline appearance and is probably a degenerative phenomenon.

Sarcoidosis. Sarcoid granulomas surrounded by dense, brightly eosinophilic lamellar collagen. Note the rosette-like adventitial distribution in this example. The central structure in this image is a pulmonary artery. H&E stain, 40× original magnification.
Regardless of specific aetiology, all granulomas can be associated with distinctive inclusions, such as the haematoxyphilic Schaumann body, lucent oxalate crystals and eosinophilic asteroid bodies in the cytoplasm of multinucleate giant cells. Oxalate crystals are brightly birefringent in polarised light and should not be confused with foreign material or a pneumoconiosis.
5b: nodules with macrophages and dust
The nodules of silicosis and silicatosis tend to be round and variably fibrotic. The fibrosis may have a whorled, lamellar or hyaline character, and almost invariably there will be admixed polarisable silicates (aluminium and magnesium salts of silica).40 Like sarcoidosis and lymphangitic neoplasms, many pneumoconioses will have a lymphatic pattern (disease occurring along bronchovascular bundles, interlobular septa and pleura) when observed at scanning magnification (fig 26). Pathologists rarely make the diagnosis of pneumoconiosis on lung biopsy specimens, as this must be performed within the clinical and radiological context.

Silicatosis. Similar to sarcoidosis in distribution, the nodules of silicate disease can be distinguished by the common occurrence of dark pigment dust and the characteristic fibroblastic proliferation that occurs in response to silicate dust. When these morphological features are present, refractile silicate particles that rotate plane-polarised light are always present. This was a patient with mixed dust pneumoconiosis. H&E stain, 40× original magnification.
5c: nodules with Langerhans cells and stellate scars
The nodular lesions of LCH have a mixed composition, including fibroblasts, collagen, Langerhans cells and pigmented alveolar macrophages.24 The nodular phase of LCH can be sufficiently cellular to suggest neoplasm (fig 27). In my experience the nodules of LCH often coexist in the biopsy specimen with variably cellular stellate scars, and these tend to have fewer Langerhans cells than the nodular form.

Langerhans cell histiocytosis (LCH). LCH has variable morphology. The cellular form can suggest neoplasm (A). At higher magnification, the characteristic admixture of pale amphophilic Langerhans cells, lightly pigmented macrophages and eosinophils confirms the diagnosis (B). (A,B) H&E stain; (A) 40× original magnification; (B) 400× original magnification.
PATTERN 6: MINIMAL CHANGES
Basic elements of the pattern: little evident pathology at scanning magnification (fig 28)

Pattern 6: minimal changes. Pattern 6 is defined by little evident pathology at scanning magnification. This patient has constrictive bronchiolitis and was severely hypoxic. There should be a bronchiole for nearly every pulmonary artery in this field (and the pair should be of nearly equal diameter in cross-section). H&E stain, 15× original magnification.
Key modifiers: with constrictive bronchiolitis, with vascular or lymphatic pathology, with cysts.
6a: minimal changes with constrictive bronchiolitis
The lung biopsy sample from a patient with clinical evidence of ILD may appear normal.41 A “normal” biopsy specimen in a patient with clinical evidence of ILD should lead to a review of the clinical and HRCT findings. Some of the conditions associated with pattern 6 may present with more dramatic findings in the biopsy specimen, but these are the diseases that can have subtle pathology and cause confusion for the histopathologist. Chronic passive cardiac congestion and pulmonary veno-occlusive disease may manifest as ILD. Early pulmonary oedema or early diffuse alveolar damage may feature endothelial vacuolisation, lymphatic dilatation and interstitial widening. Embolic diseases (eg, fat, fibrin) should be considered in the appropriate clinical setting.
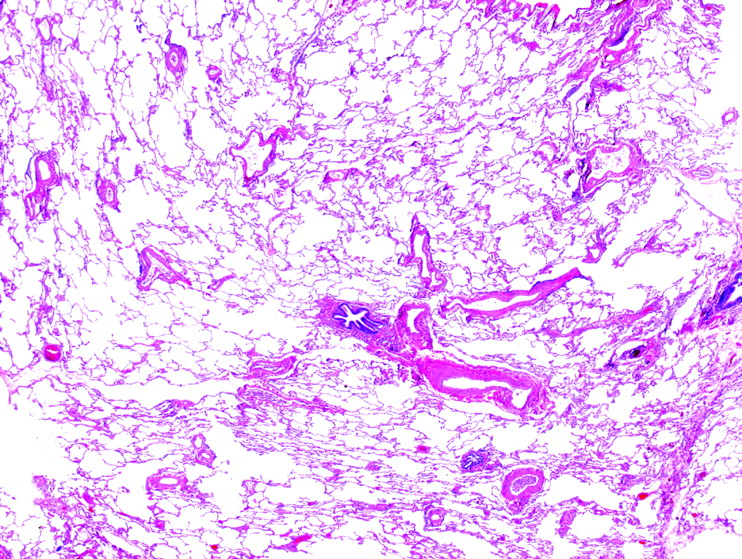
Pathological changes in constrictive bronchiolitis may be quite subtle under the microscope, despite significant clinical and radiological evidence of ILD (fig 29). Changes include a decrease in airway lumen size or complete obliteration of terminal airways to a variable degree, muscular hypertrophy, submucosal fibrous thickening, mild chronic inflammation, ectasia with mucostasis, peribronchiolar scarring and metaplastic bronchiolar epithelium that extends along surrounding alveolar walls. Although most cases that show these features are associated with airflow obstruction and radiographic hyperinflation, a small and ill-defined group of patients with small-airways disease alone present with clinical ILD, clinically and radiologically indistinguishable from other ILDs.42 Inspiratory and expiratory HRCT scans may be helpful in this setting by showing mosaic attenuation in the expiratory phase images.
Constrictive bronchiolitis. The injury in constrictive bronchiolitis is often multifocal, resulting in varicosity of the terminal airways. Here a small scarred bronchiole is present next to two profiles of a recently bifurcated pulmonary artery. H&E stain, 40× original magnification.
6b: minimal changes with vascular or lymphatic pathology
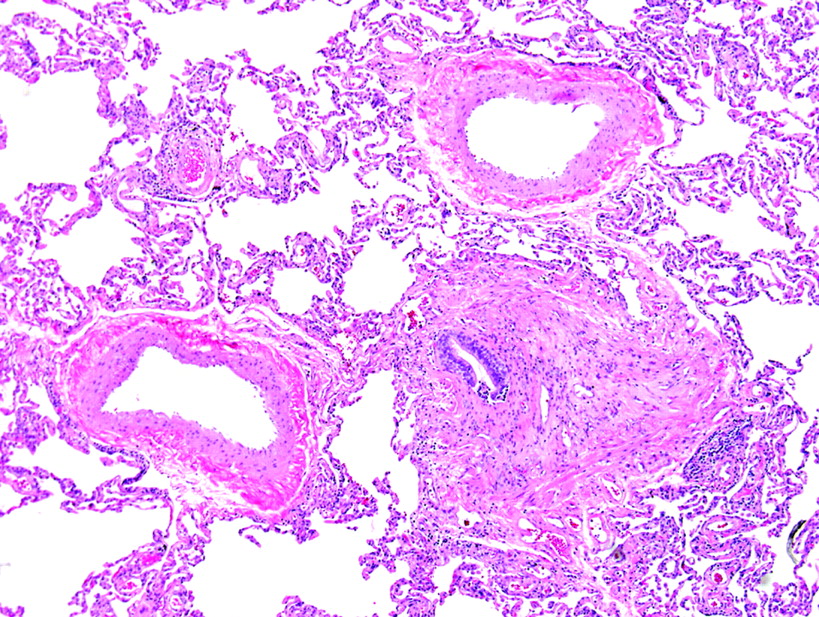
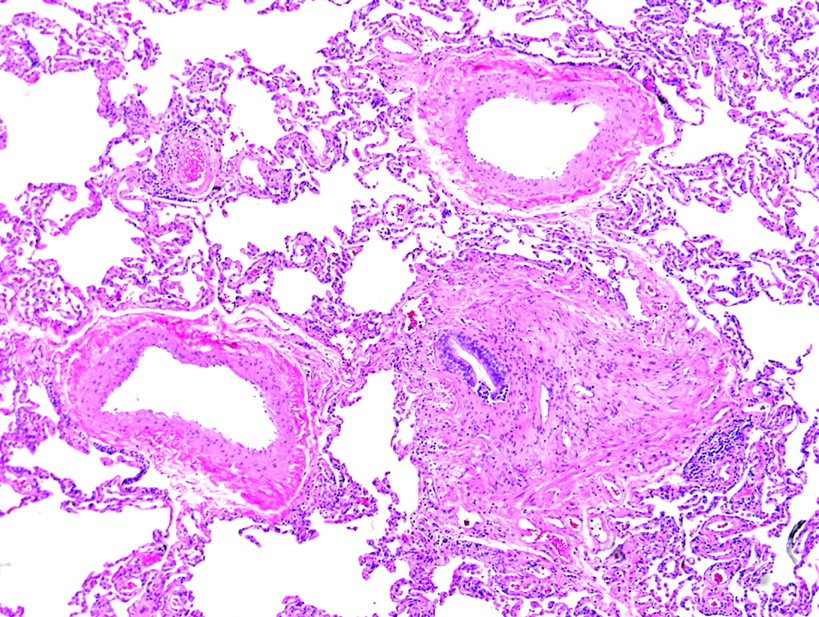
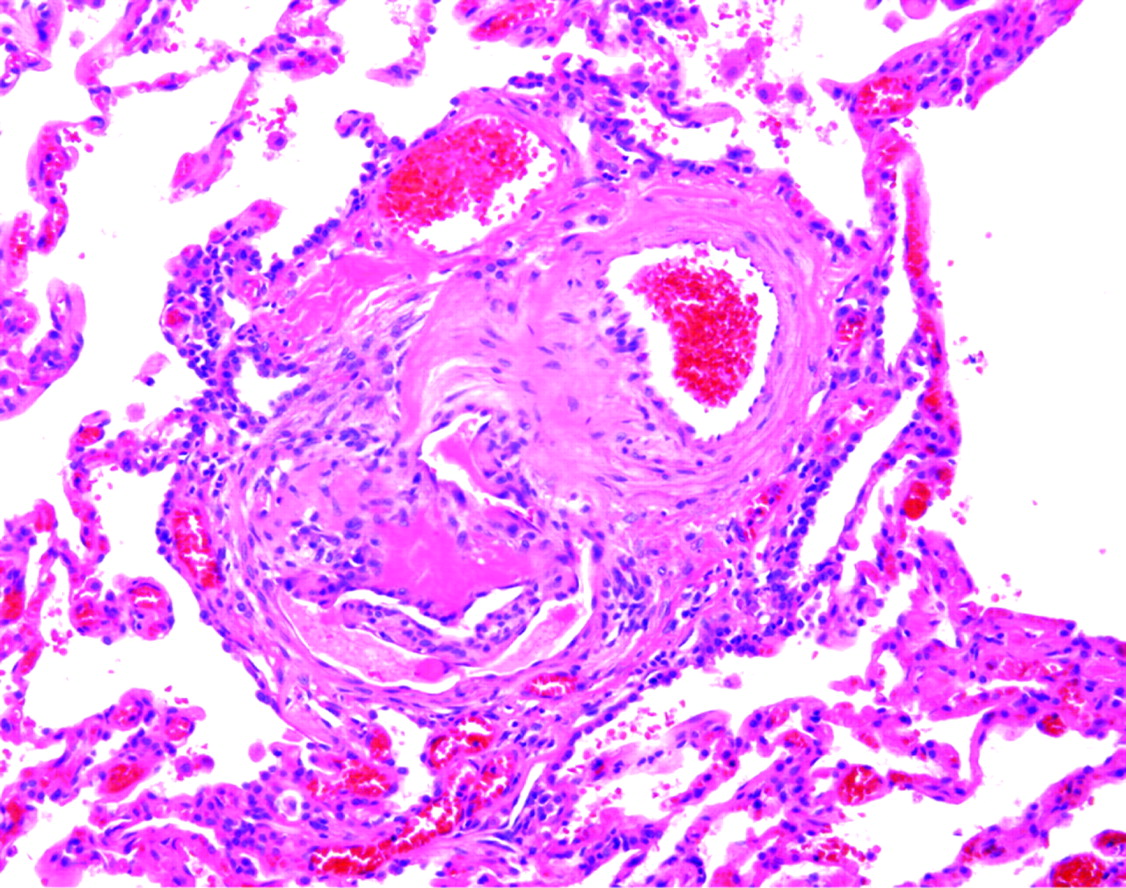
Diseases affecting the pulmonary arteries, veins and lymphatics can produce subtle changes in the surgical lung biopsy specimen. A careful and systematic evaluation of these structures is always justified when pattern 6 is encountered. When chronic disease is present in the bronchioles (such as may occur with peribronchiolar metaplasia and constrictive bronchiolitis), the adjacent arteries may become irregularly thickened and tortuous without apparent physiological consequences of pulmonary hypertension. Before considering a histopathological diagnosis of pulmonary hypertension, plexiform lesions (fig 30) or many hypereosinophilic arterioles with concentric luminal compromise (to the point of near-obliteration) should be evident.

Plexiform lesion of pulmonary hypertension. This classical plexiform lesion is composed of a pulmonary artery profile (upper right of centre) with an adjacent glomeruloid structure (lower left of centre). An early dilatation lesion is also present here (thin-walled, dilated vessels at the edges of the complex). H&E stain, 40× original magnification.
6c: minimal changes with cysts
Lymphangioleiomyomatosis is the prototype cystic lung disease which can be quite subtle in surgical lung biopsy samples. In well-prepared specimens (best achieved by removing the staples and then shaking the wedge specimen in fixative before sectioning), the cysts will be apparent at scanning magnification. Once identified, a search for thickened cellular areas of the cyst wall is often fruitful (fig 31). Immunohistochemical stains (HMB-45, Melan-A, oestrogen and progesterone receptors) are sometimes helpful in identifying the abnormal smooth muscle in this disorder.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lymphangioleiomyomatosis. The cysts of lymphangioleiomyomatosis can be quite subtle in surgical lung biopsy samples. Once a cyst is identified, a search for thickened cellular areas of the wall will reveal the aggregated fascicles of hyperchromatic and slightly disorganised smooth muscle of lymphangioleiomyomatosis (inset). H&E stain, 15× original magnification; inset: 400× original magnification.
THE TRANSBRONCHIAL BIOPSY
The pattern-based approach works also for the limited samples obtained by transbronchial biopsy.4 The size limitation here requires a somewhat more focused evaluation. The patterns of the transbronchial biopsy are presented in box 8.
A WORD ON PATTERNS OCCURRING TOGETHER
Inevitably, some overlap occurs between patterns, and this can be a useful guide to the correct diagnosis. For example, some infections are both nodular and have airspace filling (eg, botryomycosis, aspiration pneumonia), whereas others are characterised by acute lung injury and diffuse airspace filling (eg, pneumoccocal pneumonia, pneumocystis pneumonia). In fact, for some diffuse inflammatory conditions in the lung, all six patterns may be present in different areas of the same biopsy specimen (a nice example of this can be found in patients with “rheumatoid lung”).
In practice, recognising the dominant pattern is essential to navigating the differential diagnosis and addressing the primary clinical concern. As mentioned above, certain patterns should be considered dominant over others on the basis of clinical concerns. Acute lung injury (pattern 1) always trumps other patterns, given the acuity of the clinical presentation when this pattern is present and the potentially lethal immediate consequences. For example, if the biopsy specimen shows alveolar fibrin with areas of alveolar filling by OP, the successful student of the six-pattern approach would report the diagnosis as “Acute fibrinous lung injury with organisation. Special stains for organisms are negative. The differential diagnosis includes infection, toxic reaction to drug or medication, an acute manifestation of systemic autoimmune disease in the lung, and an idiopathic form.”
SUMMARY
An organised approach to the diagnosis of ILD relies on six basic histopathological reaction patterns. Additional microscopic features help to narrow the differential diagnosis. A pattern-based histopathological approach is enhanced and made more relevant with knowledge of the patient’s clinical and radiological patterns of disease.
Acknowledgments
I am indebted to Dr Thomas V Colby, whose mentoring and friendship over many years has made this work possible, and to Margaret Leslie for inspiration, support and invaluable assistance with the manuscript.
REFERENCES
Footnotes
▸ An additional table is published online only at http://jcp.bmj.com/content/vol62/issue5
Competing interests: None.