Article Text

Abstract
Background Colorectal cancer patients harbouring KRAS mutations in codon 12 or 13 do not benefit from current anti-epidermal growth factor receptor (EGFR) monoclonal antibody therapies. Efficient and robust methods are therefore required for routine clinical testing of KRAS mutation status.
Aims To evaluate a novel multiplex assay for the rapid detection of common KRAS mutations in formalin-fixed paraffin-embedded (FFPE) tissues.
Methods Genomic DNA was amplified by multiplex PCR using primers targeting the KRAS codon 12/13 region and an internal control gene. PCR products were hybridised on a liquid bead array containing target-specific probes and detected by particle flow cytometry.
Results Analytical performance assessed with plasmid DNA and genomic DNA extracted from cell lines or model FFPE cell line dilutions showed specific detection of seven distinct KRAS mutations with a limit of detection equivalent to 1% tumour. The assay was evaluated at two independent sites with a total of 140 clinical specimens. At site 1, about 45% of the specimens from a set of 86 archived FFPE blocks with unknown KRAS mutation status were found positive for a KRAS mutation. At site 2, each of the seven mutations was detected in at least five independent specimens from a selected set of 54 residual genomic DNAs previously tested with an ARMS/Scorpion laboratory-developed test.
Conclusions This novel single-well assay is a sensitive tool compatible with the clinical laboratory workflow for the rapid assessment of KRAS mutations in solid tumour specimens. Its performance and multiplex format warrant the development of broader panels including other relevant mutations in the EGFR pathway.
- KRAS
- mutation
- EGFR
- colorectal cancer
- FFPE
- diagnostics
- molecular pathology
Statistics from Altmetric.com
Introduction
The epidermal growth factor receptor (EGFR) controls a complex and critical signalling network that regulates cell growth, migration, differentiation, adhesion and apoptosis. Because this pathway is often activated in multiple cancer types, several treatment regimens based on EGFR-targeted therapies have been developed.1 2 In colorectal cancer (CRC), two monoclonal antibodies (mAbs) designed to block the binding of EGFR-specific ligands have shown consistent response rates, alone or in combination with conventional chemotherapy.3–6 Both cetuximab and panitumumab are IgG mAbs approved in Europe and in the USA for the treatment of stage IV metastatic CRC. Recently, genetic analyses have shown that specific mutations in effector genes downstream of EGFR, such as KRAS, correlate with a lack of response to these mAb therapies.7–11 These findings have significantly affected the standards of care for metatstatic CRC patients in Europe and in the USA by enabling an enrichment of the population more likely to benefit from cetuximab or panitumumab treatment.12 13
KRAS is a GTPase that plays a key role in the EGFR pathway by activating downstream effectors. Mutations in KRAS have been observed at a relatively high frequency in a variety of cancer types and in ∼40% of CRC cases.12 14 In cancer cells, most mutations impair the GTPase activity and result in the accumulation of the active GTP-bound conformation of the enzyme. This constitutively activated form of KRAS negates the upstream therapeutic blockage of EGFR by mAb.9 15 Over a dozen clinical studies have convincingly demonstrated that response to anti-EGFR mAb therapies is restricted to patients harbouring tumours wild-type for KRAS codons 12 and 13.12–14 Although many different KRAS mutations have been reported in the literature, the vast majority of activating KRAS mutations (>98%) are found within these two codons.7 8 14 By selecting the tumour specimens suitable for analysis and by ensuring that patients are screened for the appropriate KRAS mutations using a test method with the required performance characteristics, pathologists play a critical role in the management of metastatic CRC.13 14
Many KRAS testing methods compatible with formalin-fixed, paraffin-embedded (FFPE) CRC specimens have been reported in the literature and/or are commercially available.13 16 Methods commonly used in clinical settings include gene sequencing or mutation detection by real-time PCR. Direct gene sequencing is in general performed by pyrosequencing (sequencing by synthesis) or by the standard dideoxyribonucleotide (ddNTP) chain termination method.17 Real-time PCR-based technology platforms include high-resolution melting analysis or allele-specific amplification with bifunctional fluorescent primer/probe detection (ARMS/Scorpion).7 18–20 Other multiplexed mutation-specific methods, such as end-point PCR followed by multiplex single-nucleotide probe extension and size fractionation, have also been developed and successfully implemented in clinical laboratories.10 21
The recent and urgent need for efficient and robust methods for KRAS routine clinical testing has resulted in multiple studies specifically designed to assess and compare the various technologies and commercial kits available.16 22–25 Although these reports concluded that most, but not all, methods are suitable for KRAS mutation analysis in clinical specimens, they also pointed to significant differences and specific limitations in terms of sensitivity, specificity, overall performance, cost, turnaround time or workflow. We report here the evaluation of a novel method for the analysis of KRAS mutation status that was designed and developed to combine rapid multiplex detection with a streamlined workflow optimised for the molecular laboratory and without compromising analytical specificity or sensitivity.
Materials and methods
DNA specimens
FFPE blocks from colon or lung carcinoma patients were collected as part of standard clinical care and acquired from various sources by Asuragen Inc under an IRB-approved protocol (site 1, n=86). Genomic DNA was isolated from 20 μm slices using a laboratory-validated method based on the RecoverAll kit (Ambion, a Life Technologies company, Austin, TX, USA). Residual genomic DNA collected and archived at Clarient Inc were prepared from various micro-dissected FFPE CRC specimens using a proprietary laboratory-validated method (site 2, n=54). All human specimens in this study were deidentified.
Cell lines (ATCC, Manassas, VA, USA) were cultured according to the supplier's recommendations, and genomic DNA was isolated with the QIAamp DNA Mini kit according to manufacturer's instructions for cultured cells (Qiagen, Hilden, Germany). When indicated, genomic DNA from KRAS-positive cell lines were diluted in genomic DNA from a KRAS wild-type cell line (HT-29) keeping the total concentration of genomic DNA constant.
Synthetic plasmid DNA carrying specific KRAS mutations was prepared using standard molecular biology methods, purified using the QIAprep Spin Miniprep kit (Qiagen), sequence confirmed, diluted in Tris/EDTA buffer, and subsequently diluted in HT-29 genomic DNA keeping the concentration of genomic DNA constant.
FFPE specimens consisting of KRAS-positive cells diluted at 20%, 10%, 5%, 2% and 1% in a background of KRAS wild-type cells were generously provided for evaluation use only by Acrometrix, a Life Technologies company (Benicia, CA, USA). Genomic DNA was extracted from 20 μm slices using the laboratory-validated method of site 1.
DNA specimens were quantified using a NanoDrop ND1000 (NanoDrop Technologies, Wilmington, DE, USA).
Signature assay
Up to 4.5 μl of each DNA specimen was first amplified by multiplex PCR using the provided primer mix (forward and reverse biotin-modified KRAS-specific primers), 12.5 μl of provided PCR buffers, 0.05 U uracil N-glycosylase (Epicentre, Madison, WI, USA), and 2.5 U AmpliTaq Gold (Applied Biosystems, a Life Technologies company, Foster City, CA, USA) in 25 μl reaction mixtures. Amplifications were performed in 96-well PCR plates (AB-Gene, Rockford, IL, USA) using the following programme: 37°C for 15 min, 95°C for 10 min, 45 cycles consisting of 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and hold at 4°C. Biotinylated amplified products (5 μl) were then directly hybridised to 45 μl of provided bead mix containing seven distinct capture probes specific for each KRAS mutation and covalently bound to seven distinct fluorescent microspheres. The hybridisation reactions were performed in 96-well plates for 30 min at 52°C. After hybridisation, the reaction mixtures were immediately diluted with 75 μl of provided hybridisation buffer at room temperature and centrifuged for 3 min at 1200 g. After removal of 100 μl supernatant, the 96-well plates were transferred to a Luminex 200 system (Luminex Corp, Austin, TX, USA) prewarmed at 52°C, where 50 μl of provided reporter solution containing the streptavidin–phycoerythrin conjugate were then added to each hybridisation reaction well before the start of the automatic fluorescence analysis. The median fluorescent intensity (MFI) detected by the Luminex system on at least 50 beads of each type was subsequently analysed in Excel (Microsoft Corp). The signal generated by bead ID numbers 19, 28, 37, 38, 47, 48, or 58 relative to a predetermined cut-off value was used to make a positive or negative call for G12V, G12A, G13D, G12C, G12D, G12S and G12R, respectively. For this research study, a cut-off at 500 MFI corresponding to a theoretical false-positive rate of <0.001% was selected based on the mean MFI value and SD of the normal-like distribution of background signals observed with true-negative DNA samples (data not shown). A conserved genomic sequence was co-amplified in each sample and concurrently detected on an independent bead type (ID 45) to serve as endogenous control (EC). Primer and probe sequences were according to the Catalogue of Somatic Mutations in Cancer website (http://www.sanger.ac.uk/genetics/CGP/cosmic, last accessed 22 July 2010). Additional procedural information can be found in the Signature KRAS Mutations 7 (Research Use Only) kit instructions (Asuragen Inc).
Negative and positive (G12V) control samples were processed with each plate of specimens to validate the PCR, hybridisation and detection steps in each run.
A GeneAmp PCR System 9700 (Applied Biosystems) or a Veriti 96-well Thermal Cycler (Applied Biosystems) was used for the PCR and hybridisation steps at sites 1 and 2, respectively.
Other assays
For DNA sequencing, standard PCRs were performed using a pair of primers flanking the KRAS codon 12 and 13 mutation site (Integrated DNA Technology, Coralville, IA,USA) and 35 PCR cycles (94°C for 30 s, 55°C for 30 s, 72°C for 60 s) followed by a 10 min elongation step at 72°C. Bidirectional sequencing of the PCR products using the BigDye terminator method was performed by ACGT Inc (Wheeling, IL, USA). The 54 clinical specimens from site 2 were tested for KRAS mutation status at site 2 (Clarient Inc) using an ARMS/Scorpion laboratory-developed test based on the DxS KRAS Mutation Detection kit (DxS, a Qiagen company).
Results
The Signature KRAS Mutations 7 assay (Research Use Only) is a qualitative assay for the simultaneous detection of seven common KRAS mutations (G12A, GGT→GCT; G12C, GGT→TGT; G12D, GGT→GAT; G12R, GGT→CGT; G12S, GGT→AGT; G12V, GGT→GTT; G13D, GGC→GAC) and an EC gene. After genomic DNA extraction using standard laboratory-validated methods, the EC and KRAS codon 12/13 gene regions are amplified by multiplex PCR in a single well of a 96-well plate. The PCR products are then transferred to a second plate where they are hybridised to target-specific capture probes bound to spectrally distinct beads. After addition of a reporter molecule, the MFI generated by each bead is determined by microsphere flow cytometry. If the MFI of a given KRAS mutation bead is greater than a fixed cut-off (500 MFI for this study), the specimen is reported positive for that specific mutation. The assay includes positive and negative controls to assess the validity of each run and is performed in about 4 h with about 45 min of hands-on time for a 96-well plate.
The assay analytical specificity was evaluated using genomic DNA isolated from cell lines wild-type or positive for specific KRAS mutations and confirmed by sequencing. Mutations G12A, G12C, G12D, G12S, G12V and G13D were specifically detected in six independent cell lines (figure 1). No signal above the cut-off was observed with a cell line wild-type for KRAS (HT-29). An input mass titration experiment also showed that each mutation was reproducibly detected by the assay in 1–20 ng genomic DNA (online supplementary table 1). Specificity was confirmed using individual plasmid DNA carrying each of the six mutations (data not shown) and a plasmid carrying the codon 12 GGT to CGT mutation (G12R; figure 1). Analytical specificity was further confirmed using at least five independent clinical specimens positive for each of the seven KRAS mutations (see below).
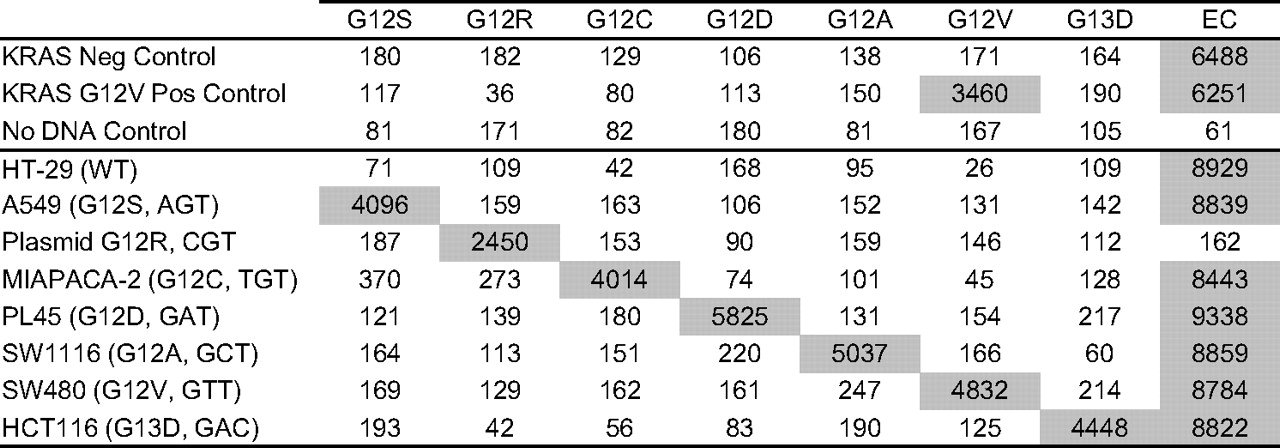
Analytical specificity. Representative examples of signal (median fluorescent intensity) obtained with genomic DNA (10 ng) isolated from seven cultured cell lines. The KRAS mutation status determined by sequencing for each cell line DNA is indicated in parentheses. For the G12R-positive plasmid, ∼1500 copies were added to the PCR or the equivalent of ∼5 ng mutant genomic DNA assuming 3.3 pg DNA per human genome. Assay results obtained with the kit positive and negative controls and a no DNA control (water) are also shown. EC, endogenous control.
To assess analytical sensitivity, each of the six KRAS-positive cell line genomic DNAs was serially diluted in a background of KRAS wild-type genomic DNA provided by the HT-29 cell line. Analysis of the mean signal generated by the positive mutant probes showed that the assay can reproducibly detect KRAS mutations in a 10% or 1% genomic DNA dilution (figure 2). Individual results for each cell line are shown in supplementary figure 1A (available online). With an additional 10-fold dilution (0.1%), the assay did not reproducibly generate MFI values above the cut-off (data not shown), suggesting a limit of detection at or just below 1%. We further investigated the assay's analytical sensitivity using model FFPE cell line dilutions that more closely mimic clinical specimens with low tumour cell content (see Materials and methods). Positive signals were reproducibly observed in genomic DNA extracted from FFPE blocks consisting of three distinct KRAS-positive cells diluted at 20%, 10%, 5%, 2% or 1% in a background of KRAS wild-type cells (figure 2; see supplementary figure 1B for individual results). This experiment confirmed that the assay limit of detection is about 1%.

Analytical sensitivity. The graph shows the average signal (median fluorescent intensity (MFI)) generated by the endogenous control (EC) probe and the KRAS mutant positive probes for each dilution tested. The ‘genomic DNA’ values were calculated by testing six KRAS mutant positive genomic DNAs (G12A, G12C, G12D, G12S, G12V or G13D) undiluted (100%) or diluted in wild-type HT-29 genomic DNA (10% or 1%). The ‘FFPE cell line’ values were calculated by testing genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) blocks containing three KRAS mutant positive cells (G12C, G12V or G13D) diluted in a background of KRAS wild-type cells at 20%, 10%, 5%, 2% or 1%. The error bars show the respective SDs from the mean signal for each dilution tested.
The assay was next evaluated at two independent sites using a total of 140 residual clinical specimens (figure 3). At site 1 (Asuragen), 86 archived FFPE blocks from colon (n=74) or lung (n=12) carcinoma with unknown KRAS mutation status were analysed. The genomic DNA was extracted from 20 μm slices using a laboratory-validated method and tested with the Signature assay at fixed input (10 ng) and by ddNTP sequencing as the reference method. At site 2 (Clarient), 54 residual genomic DNAs extracted from micro-dissected FFPE CRC specimens with known KRAS mutation status determined as part of routine clinical testing were analysed. The set was enriched for known positive specimens so that each of the seven KRAS mutations would be represented by at least five independent clinical specimens. Each genomic DNA was tested in duplicate at fixed volume (2 μl) with the Signature assay, and the results compared with the clinical data acquired using an ARMS/Scorpion laboratory-developed test (LDT).

Experimental design to assess the performance of the Signature KRAS Mutations 7. Eighty-six consecutive DNA specimens were compared with dideoxyribonucleotide triphosphate (ddNTP) sequencing at site 1. Fifty-four residual DNA specimens were compared with an ARMS/Scorpion laboratory-developed test (LDT) at site 2. FFPE, formalin-fixed paraffin-embedded.
All genomic DNAs were successfully extracted from the 86 archived FFPE blocks and tested at site 1 (table 1). About 47.3% (35/74) of the colorectal carcinoma specimens and 45.3% (39/86) of the total set were found to be positive for a single KRAS mutation using the Signature assay (see online supplementary table 2 for a complete list of results by specimen type). The mutations G12D, G12V and G13D were most often identified and represented about 33.3% (13/39), 25.6% (10/39) and 20.5% (8/39) of the positive cases, respectively (table 1). Non-target negative MFI signals were low and reproducible at least twofold below the 500 MFI cut-off. Representative examples of assay results using 10 or 2 ng genomic DNA input for each mutation detected at site 1 and one negative specimen are shown in figure 4. Comparison with sequencing data resulted in 90.6% (77/85) overall agreement (table 2). No sequencing results could be obtained for one Signature-negative specimen, and about 20.5% (8/39) of the specimens positive with the Signature assay were found negative by sequencing.
Summary of KRAS mutation status in 140 independent clinical specimens as determined with the Signature KRAS Mutations 7 assay at two sites
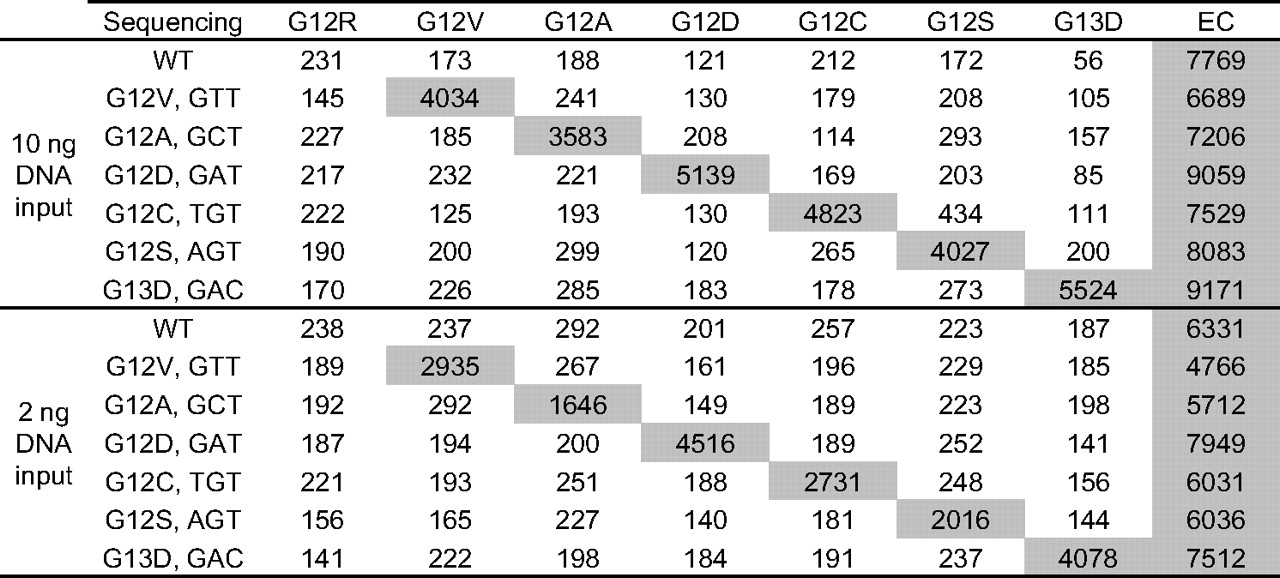
Signature KRAS Mutations 7 results at site 1. Representative examples of signal (median fluorescent intensity) obtained with genomic DNA extracted from archived formalin-fixed paraffin-embedded blocks at site 1. DNA concentration was normalised, and a fixed input mass (10 or 2 ng) was used per PCR. KRAS mutation status as determined by sequencing is shown on the left. EC, endogenous control; WT, wild-type.
Summary of Signature KRAS Mutations 7 assay performance versus sequencing for 85 specimens at site 1
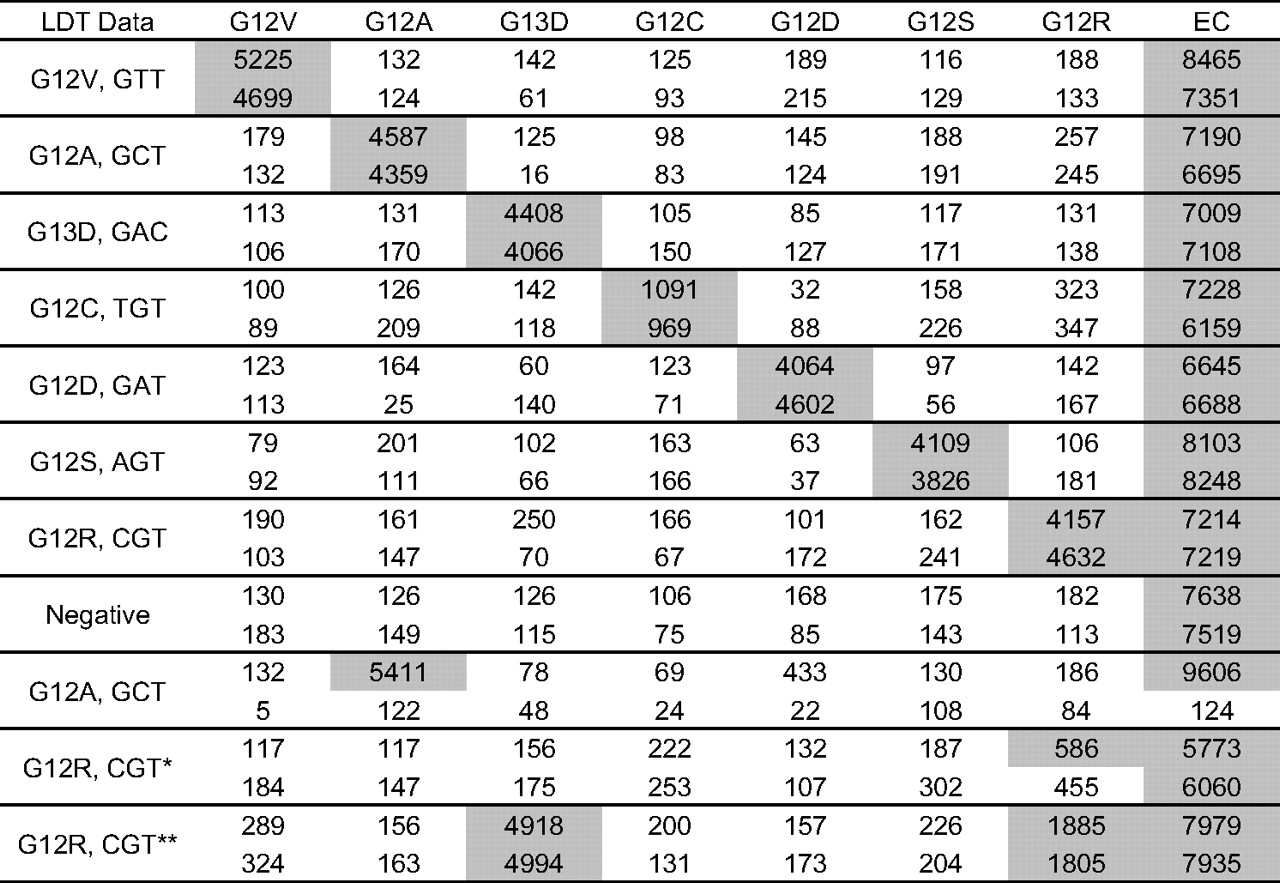
The 54 residual genomic DNAs archived at site 2 were also successfully tested with the Signature assay. Each KRAS mutation was detected among the 43 KRAS-positive specimens selected (table 1). Representative examples of duplicate testing for each mutation and a negative specimen are shown in figure 5. There was excellent agreement between the replicates with two exceptions. One G12A positive specimen did not generate any signal in one of the two replicates, and the absence of EC signal resulted in a fail call. Another specimen generated signal above the cut-off (500 MFI) in only one of the two replicates. This specimen was a weak G12R positive with the reference method and the Signature assay (455 and 586 MFI) and was later found to be negative by sequencing (see below). Overall, there was 100% positive and negative agreement between the Signature assay and the LDT at site 2 (table 3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Signature KRAS Mutations 7 results at site 2. Representative examples of signal (median fluorescent intensity) obtained with various input of residual genomic DNAs extracted from clinical specimens at site 2. DNA concentration was >3 ng/μl, and a fixed volume input was used (2 μl). KRAS mutation status as determined by an ARMS/Scorpion laboratory-developed test (LDT) is shown on the left. *Weak positive by LDT and Signature, negative by sequencing. **Double positive by Signature, confirmed by sequencing.
Summary of Signature KRAS Mutations 7 assay performance versus an ARMS/Scorpion laboratory-developed test (LDT) for 54 specimens at site 2
A subset of 34 residual genomic DNAs from site 2 was also tested by sequencing (see online supplementary table 3 for a summary of results for each specimen). There was complete agreement between all three methods for about 80% (27/34) of the specimens. One specimen reported as G12R positive by the LDT was found double positive for G12R and G13D by sequencing and with the Signature assay (figure 5). Among the cases reported KRAS positive by both the Signature assay and the LDT, about 20.7% (6/29) were found wild-type by sequencing (table 4), similar to the fraction observed at site 1 (8/39 or 20.5%; see table 2). We concluded that the Signature assay has an excellent performance relative to methods with similar analytical sensitivity and is an appropriate tool for the detection of KRAS mutations in FFPE clinical specimens.
Summary of Signature KRAS Mutations 7 assay performance versus sequencing for 34 specimens at site 2
Discussion
The discovery of activating mutations in the KRAS gene has enabled the rapid adoption of individualised treatment regimens for metastatic CRC patients. The identification of patients most likely to not respond to anti-EGFR mAb is, however, dependent on the accurate and sensitive detection of specific KRAS mutations. The Signature KRAS Mutations assay is a research tool that was designed and developed to answer the current clinical needs for improved detection methods with appropriate mutation coverage, analytical performance and laboratory efficiency. The 96-well plate assay consists of reagents for multiplex PCR and hybridisation/detection of seven relevant KRAS mutations in codons 12 and 13 and an endogenous control sequence. The assay requires additional components that are already commonly found in many clinical laboratories: reagents for DNA extraction from FFPE specimens, a standard 96-well thermocycler, and the Luminex 100 or 200 flow cytometer. This platform is compliant with 21 CFR part 11 and is compatible with multiple nucleic acid- or protein-based tests already approved by the Food and Drug Administration (http://www.fda.gov, last accessed 9 May 2010). Multiplex detection of seven KRAS mutations in a single well of a 96-well plate is expected to greatly improve operational efficiency and to potentially decrease the cost of current commercial singleplex real-time PCR assays by up to 50%. As with any sensitive PCR-based method, the risk of potential false-positive results should be controlled by following standard good laboratory/clinical practices and a unidirectional workflow. The risk of cross-contamination can be further mitigated by inclusion of uracil N-glycosylase and appropriate negative controls in every run. Overall, the Signature assay design is compatible with the molecular laboratory workflow, and we undertook to evaluate its analytical and clinical performance for the testing of FFPE specimens.
The Signature assay is sequence-specific and was designed to detect seven activating KRAS mutations that have been clinically validated and represent over 98% of all KRAS mutations.7–10 15 Many other KRAS mutations have been described in the literature (http://www.sanger.ac.uk/genetics/CGP/cosmic, last accessed 22 July 2010), and it should be noted that a negative assay result does not rule out the presence of potential other rarer KRAS mutation(s) that may or may not hinder effective anti-EGFR treatment. To increase the clinical sensitivity of the assay, additional mutations relevant in metastatic CRC, such as G13C, G13S or G13R, would have to be included in the panel. Such an assay has been developed and successfully tested with clinical specimens (unpublished work). The analytical specificity of the current assay was confirmed by testing plasmid DNA and genomic DNA from well-characterised cell lines and independent clinical specimens (figures 1, 4 and 5). For this type of qualitative assay, data interpretation is simple, and specificity was demonstrated by the reproducible detection of positive and negative signals above or below a fixed cut-off value (500 MFI). Importantly, none of the specimens confirmed negative by independent methods generated MFI signals above the cut-off in this study. False-positive rates as high as 20% have been reported for other screening methods, which is not acceptable for a negative predictor such as KRAS where the burden of a false positive is consequent.23 Variability in background negative signals can, however, be observed with the Signature assay according to the preanalytical steps or the level of training of the operator (data not shown). The qualitative cut-off must therefore be appropriately validated by testing representative true-negative specimens and verifying the distribution of expected background negative signals. Having a simple qualitative data interpretation method based on a positive cut-off further enables monitoring and potentially adjustment of the cut-off after changes to the technique, the DNA extraction method, or the laboratory-specific testing algorithm.
The distribution of positive signal obtained with the Signature assay can in theory cover a broad range of MFI according to the relative abundance of tumour cells in the starting specimen and/or the DNA input per PCR. Most clinical specimens tested in this study generated robust MFI at least 4–5-fold above the cut-off (figures 4 and 5). To evaluate analytical sensitivity, serial dilution experiments were performed using genomic DNA extracted from various cell line models. Our results indicate a limit of detection of about 1% tumour cells (figure 2). It is important to note, however, that our reported sensitivity does not correspond to the absolute analytical sensitivity. Cell lines often exhibit alterations in gene copy number that can further vary during cell culture passages and the exact number of KRAS gene copy was unknown at the time of DNA extraction.13 These variations in copy number may explain why specific cell lines at 1% dilution resulted in mean signal around 500 MFI, while others generated higher MFI, 3–5-fold above the cut-off (online supplementary figure 1). The same pattern was also observed when testing the assay input range with robust signals varying between 1000 and 4000 MFI in as low as 1 ng input (online supplementary table 1). Considering that the mass of a haploid human genome is about 3.3 pg, 1 ng of cell line DNA would correspond to about 300 copies, and a 1% dilution at 20 ng input to about 200 pg of mutant DNA or 60 copies. Therefore, even if multiple copies of the KRAS genes were present in our cell lines, the absolute analytical sensitivity of the assay is likely to be around 100–200 copies of mutant KRAS sequence.
Our study was not designed to directly compare various KRAS testing methods in terms of performance or workflow. Rather, our goal was to establish the analytical performance of the assay and to validate its potential clinical utility using two independent reference methods with known performance characteristics.16 22–25 The testing of 140 representative clinical specimens brought some valuable insights into the relative performance of these methods. At both sites, about 20% of the specimens found positive by Signature were negative by sequencing. These specimens were likely false negative as they were confirmed positive by an independent LDT at site 2 (online supplementary table 3). Direct gene sequencing by the ddNTP termination method has long been considered the ‘gold standard’ for the identification of gene mutations. Unlike sequence-specific assays such as the Signature assay, this method can in theory detect all possible mutations in codons 12 and 13. However, its reported analytical sensitivity is only 10–20%, which can partially explain the high false-negative rate observed in our study.17 This problem is further enhanced when working with FFPE tissues specimens which can result in poor DNA quality and small fragmented DNA templates suboptimal for the amplification of the 100–150 bp PCR products required for direct sequencing.13 25 Even though enough tumour cells and genomic DNA can in general be recovered from clinical CRC specimens, molecular methods with high analytical sensitivity are more likely to detect KRAS-positive cells in a variable background of KRAS-negative tumour cells and can be more robust, therefore increasing the confidence in negative test results. For example, pyrosequencing may be a method better suited for FFPE specimens, as it is compatible with shorter DNA templates (60–70 bp) and has a reported sensitivity of 5–10%.17
To further improve analytical sensitivity, various methods based on real-time PCR platforms have also been developed for the detection of KRAS mutations. A technology commonly used in clinical settings is high-resolution melting analysis (HRM) with either fluorescently labelled probes or DNA intercalating dyes.19 20 This method has a reported analytical sensitivity of 5% and can detect multiple mutations in a single reaction; however, it does not directly identify the specific mutations and has a high frequency of false positive.20 23 Real-time allele-specific PCR or amplification refractory mutation system (ARMS) linked to a unique bifunctional fluorescent primer/probe molecule (Scorpion) is specific for individual mutations and has been used successfully in metastatic CRC clinical trials.7 There was 100% agreement (95% CI 93.4% to 100%) between the Signature assay and a LDT based on this technology at site 2. The ARMS/Scorpion method has an excellent analytical sensitivity (1%), but requires the set up of independent reactions for each of the seven mutations, which can limit the number of specimens tested per run or the potential requirement to expand the panel of mutations tested.13 18 23
Recent genetic studies suggest that additional activating mutations in the KRAS gene or in other downstream effector genes such as BRAF or PIK3CA are also associated with the lack of response to anti-EGFR mAb and/or poor prognosis.11 12 20 21 To test multiple mutations and multiple genes in a single reaction, mutation-specific multiplex primer extension assays have been developed and successfully used in clinical studies.10 21 These assays are more sensitive than sequencing and can detect up to 22 mutations in a single test, but require multiple steps from PCR amplification to primer removal, multiplex single-nucleotide primer extension (SNaPshot), shrimp alkaline phosphatase treatment, enzyme inactivation, size fractionation by capillary electrophoresis, and peak size analysis.10 21 A multiplexed Luminex-based detection method for KRAS mutations has also recently been described.26 Although the results were not directly compared against well-characterised reference method(s), Wu et al showed detection of two KRAS mutations, G12D and G13D, in serum samples from patients with non-small cell lung cancer and in FFPE samples from CRC patients.26 They also reported an analytical sensitivity of 100 copies by testing plasmid DNA carrying KRAS mutations.26 This modified mutant-enriched PCR method may therefore have a performance equivalent to the Signature assay. One major difference is the protocol consisting of a first PCR with mismatched primers, a digestion of the amplified wild-type DNA with a restriction enzyme, and a second nested PCR of the amplified mutant DNA with biotinylated primers before hybridisation on capture probes and detection on the Luminex flow cytometer.
Although more work is required to clinically validate the Signature KRAS Mutations assay, the data presented here demonstrate that a streamlined multiplex workflow with simple data interpretation involving only three steps (PCR, hybridisation, detection) can be achieved without compromising analytical performance. Experiments have also shown that the Signature assay can be combined with multiplex detection of five additional KRAS mutations in codon 13 (G13A, G13C, G13R, G13S and G13V) and BRAF V600E (unpublished work). The validation of broader panels including other relevant mutations in the EGFR pathway would speed up the molecular characterisation of FFPE tumour specimens and likely facilitate the development of optimal personalised treatment approaches for metastatic CRC and other cancer types.
Take-home messages
The identification of specific mutations in the KRAS gene plays an important role in colorectal cancer management.
Detection methods compatible with FFPE tumour specimens, optimised for the clinical laboratory workflow, and with the appropriate accuracy and analytical sensitivity are required.
This study shows that rapid multiplex detection of seven relevant KRAS mutations in a single-well assay can be achieved with about 1% analytical sensitivity.
Accuracy relative to existing reference methods was demonstrated by testing 140 representative clinical specimens.
References
Supplementary materials
Web Only Data JCP.2010.081539
Footnotes
Competing interests WLW, FY, ZY and EL are employees of Asuragen Inc. VT, RW, KB and PC are employees of Clarient Inc.
Provenance and peer review Not commissioned; externally peer reviewed.