Article Text

Abstract
The complexity involved in the histological interpretation of lymph nodes and other lymphoid tissue specimens suspected of harbouring lymphoma is underappreciated. As with other histology specimens, the quality of sections and background information are crucial but so, increasingly, is the appropriate use of immunocytochemistry and a variety of molecular analyses. Within the UK National Health Service, progressive regional centralisation is ongoing, to ensure access to specialist expertise and a full range of testing beyond traditional stains. This is to be welcomed but there remains a need to maintain skills in smaller district hospitals, to ensure lymphoma recognition in unexpected circumstances, to permit clinically useful interim diagnoses when needed urgently and to sustain training in haematopathology among junior pathologists. In this review a range of potential pitfalls in lymphoid tissue pathology is outlined, arising at all stages from specimen preparation to reporting. Knowledge of such pitfalls, some of which are common while others are rare but of vital clinical importance, should help increase confidence in lymphoma diagnosis among histopathologists.
- Lymph nodes
Statistics from Altmetric.com
Sources of error in haematopathology are predominantly of an administrative, clerical, technical or diagnostic nature, as in other areas of pathology. Administrative and clerical errors relate to loss, transposition and delays affecting specimens and the information relating to them. Such errors may occur at any stage from the intention to obtain a specimen to the final incorporation of reported results into a patient's record. They are of great importance, and regularly audited systems should be in place to minimise their occurrence. However, as these types of error are not specific to haematopathology, they are not discussed further in this article. It should be noted that requirements for centralised reporting/review and production of integrated reports (with information from a variety of laboratory sources—haematology, cyto and histopathology, cytogenetics, molecular genetics) place important demands on the reporting side of this equation in haematopathology.
In 2006, the National Institute for Health and Clinical Excellence issued improving outcomes guidance (IOG) for haematological cancers, within which the crucial need for a central (regionally based) specialist review of haemato-oncology diagnoses was emphasised.1 This component of the IOG is strongly influenced by audits, particularly that conducted in Wales,2 showing rates of discordance in the region of 20% between initial non-specialist and subsequent specialist diagnoses of lymphomas. In 8% of cases, the discrepancies required clinically significant changes in patient management following specialist diagnosis. The true situation throughout England and Wales before the National Institute for Health and Clinical Excellence issued this IOG was probably more complex than such audits imply, because longstanding local arrangements for regional referral of lymphoma cases for expert diagnosis pre-dated these studies in most areas of the UK. Many apparent discrepancies relate to intentionally incomplete initial diagnoses in anticipation of additional technical and diagnostic input from a regional referral centre or expert colleagues.3–5 However, the potential for selective referral and delay in final diagnosis arising from such informal arrangements was considerable. The 8% figure, representing discordance requiring a significant change in clinical management, is probably accurate. Investment in systems to ensure the completeness and timeliness of referral of haemato-oncology specimens for specialist diagnosis is currently still ongoing throughout the NHS. Post-IOG audits are awaited to indicate how regional centralisation has influenced the proportion of significant diagnostic errors.
The risk of diagnostic error and the requirement for expert review are perceived as being lower in other subspecialist areas of pathology,3 6 and the high concern in haematopathology requires explanation. Complexity of diagnosis in this field is underappreciated. There is a requirement for testing beyond examination of H&E-stained sections in almost all cases,7 and lack of access to an adequate range of immunohistochemistry in smaller hospitals is a major factor. In many examples, histopathologists are not unaware of the differential and likely final diagnosis but, crucially, lack access to the tests necessary to confirm their impressions. Also, additional testing by fluorescence in-situ hybridisation (FISH) and PCR is mainly available only in larger regional centres and is required to confirm an increasing proportion of haemato-oncological diagnoses. Lack of access, lack of familiarity and the perceived reluctance of trusts to pay for these investigations currently lead to under-use.
These sources of error are fairly obvious but a change of culture is required to prevent them. For example, it has been a common view that ‘straightforward’ Hodgkin lymphoma (HL) and low-grade follicular lymphoma can be diagnosed adequately without immunohistochemistry. However, this approach carries risk. Diagnostic ‘drift’ is likely over time without confirmation by demonstrating an appropriate immunophenotype, until there may be failure to appreciate that a case is not, in fact, straightforward at all. This is particularly likely in a smaller hospital environment in which lymphoid tissues suspected of harbouring lymphomas form only a minor component of an individual histopathologist's workload, even if that person is the lead consultant for their department in this area of cancer diagnostics. Only by regional (or even larger scale) centralisation of haemato-oncology diagnostic services, concentrating the specimens into a few laboratories with resultant high volume throughput, can the full range of immunohistochemical analyses be made available effectively.8 Likewise, costly investment to expand the range of diagnostic tests applied (new antibodies, FISH, clonality PCR, etc) can only be efficient in large regional centres. The volume of cases for reporting in these centres self-sustains the expertise of the haematopathologists, including their interpretation of difficult immunostains and molecular tests. However, there is a risk that the experts themselves may drift diagnostically if the intensity and repetitive nature of their work lead to their failing to cross-refer or to seek further diagnostic advice for challenging cases. The capacity for double-reporting and a formal scheme for external quality assurance are needed to address this; some centres may have achieved the former but the latter is not yet in place.
Inadequate background information
Another significant contributor to diagnostic error in haematopathology comes under the category of ‘rubbish in, rubbish out’. There are few diagnoses that can be made accurately or completely without good communication between pathologists, haematologists, oncologists, radiologists and others. It is commonly the case that lymph node specimens reach histopathology departments accompanied by inadequate clinical information. The clinical details provided are frequently minimised further in being relayed via surgical teams acting as technical operators to obtain the tissue without any interest in the final result for patient management.
It is often also necessary for the results of other tests to be known, particularly from haematology and genetics laboratories. This principle underlies the IOG central tenet for integrated reporting in haematopathology; very few centres are yet compliant with this, largely due to trusts' resistance to invest in the required IT infrastructure and not as a result of lack of awareness or desire on the part of haematologists, haematopathologists and geneticists to achieve this.
Inadequate or inappropriate specimens
Traditionally, the ‘ideal’ histology specimen for lymphoma diagnosis has been an excised intact lymph node, transported rapidly to the laboratory without previous fixation. This allows immediate sampling of the material on receipt to make imprint preparations (useful for rapid cytological assessment and FISH), disperse cells into medium for flow cytometry, and freeze small pieces for subsequent nucleic acid studies by PCR. If fresh tissue is needed for microbiological analysis, this should be sent directly to the appropriate laboratory from the operating theatre, avoiding potential delay and miscommunication of requirements that may arise from relying on the transfer of tissue between laboratories.
This ideal is rarely achievable, for geographical and other logistical reasons, most lymph node specimens are dispatched from theatres in fixative solution. However, this is greatly preferable to prolonged transit without fixative. In the same way that immunohistochemistry using frozen tissue has effectively disappeared from standard diagnostic practice, the increasing use of paraffin sections for PCR-based studies reduces the direct need for frozen tissue storage. However, the opportunity for rapid immunophenotyping by flow cytometry and FISH using imprints is lost if tissue is first received in fixative. A valuable resource of frozen lymphoma tissue for potential research use is also sacrificed. Whether received fresh or in fixative solution, lymph nodes and any other large specimens suspected of containing lymphoma should be inspected on receipt in the histopathology laboratory and sliced as soon as possible, after recording measurements and a brief description of the intact specimen, to aid fixation.
Increasingly, to minimise patient discomfort and increase the speed of diagnosis, fine needle aspiration (FNA) cytology and needle biopsy of lymph nodes are being employed. FNA can be immensely valuable but only plays a limited role in screening lymph nodes (for infections, granulomas and metastatic solid tumour deposits, etc) unless samples with suspected lymphoma can be immunophenotyped by fluorescence-activated cell sorting.9–11 The preparation of cytoblocks for immunohistochemistry can partly substitute for this12 but entails delay, requires more cells, and is technically laborious to add into routine FNA processing. The range of immunophenotyping is also less useful than can be achieved by fluorescence-activated cell sorting in most cases. In general, unless clear cytological confirmation of classic HL, or of a non-Hodgkin lymphoma (NHL) whose type is already known, is gained, suspicion of lymphoma from FNA should lead to the excision of the affected node/mass.
Needle core biopsy of inaccessible masses suspected of harbouring lymphoma is also an invaluable tool. Although the sample size is limited, there may be no other practical means of obtaining diagnostic tissue and under imaging guidance the specimens obtained can be quite substantial. For speed, with pressure on surgical operating lists, increasing use is being made of needle core biopsy to investigate superficial lymphadenopathy. However, under these circumstances, the limited nature of the sample is highly undesirable and this trend should be resisted by multidisciplinary teams on behalf of their patients' best interests. Even when the sample is sufficiently generous for full paraffin section immunophenotyping, it is frequently the case that there is insufficient for FISH and PCR.
Box 1 highlights common diagnostic errors in lymph node diagnosis and box 2 summarises the errors that are most critical for correct patient management.The selection of particularly important errors highlighted in box 2 reflects categories in which potentially major differences in treatment will result from a misdiagnosis.
Common situations involving risk of diagnostic error
Differential diagnosis of reactive conditions versus neoplastic
Incomplete lymphoma diagnosis
Inaccurate and misleading use of classification terminology
Incorrect assessment of grade in NHL
Classic HL versus NHL
Incorrect assessment of lymphoid infiltration at non-nodal sites
Critical diagnostic errors in lymphoid tissue diagnosis
Reactive versus neoplastic conditions
Diffuse large B cell lymphoma versus Burkitt lymphoma
Low grade versus high grade in small lymphoid cell infiltrates
Classical HL versus NHL
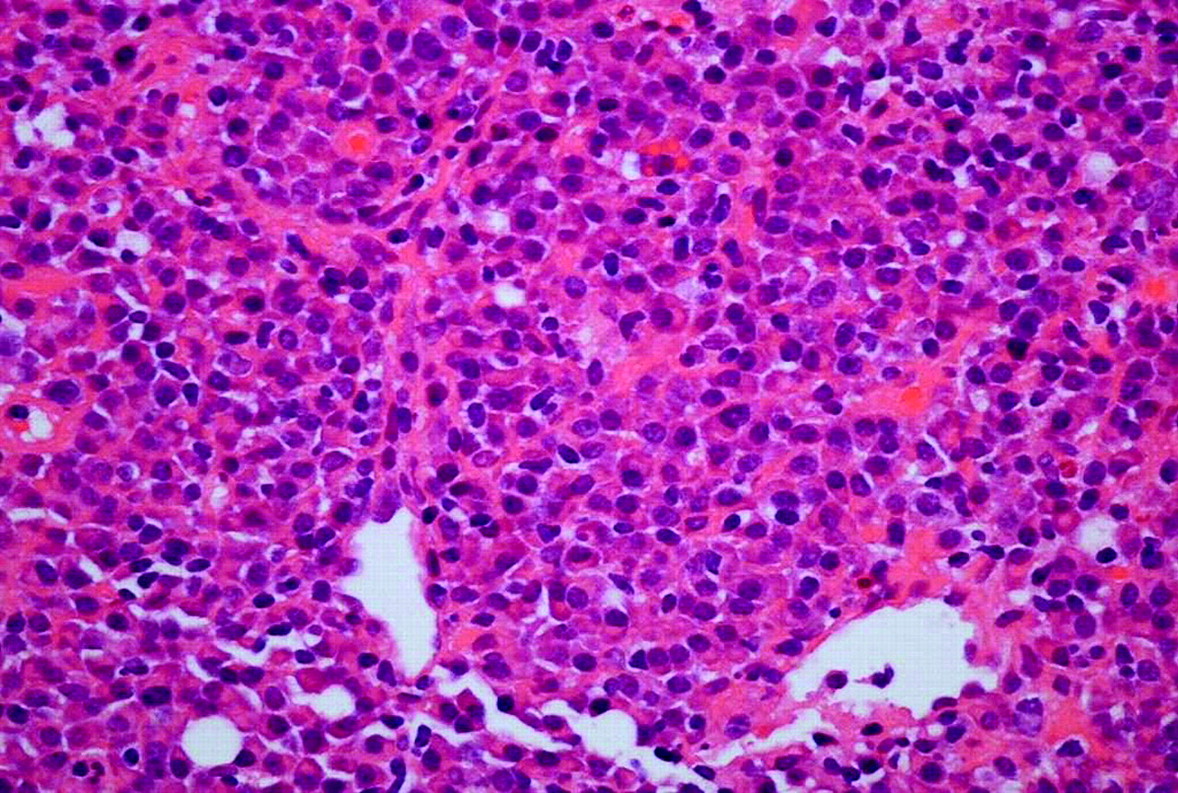
Misdiagnosis of a reactive condition such as neoplastic, or vice versa, may be disastrous for the patient, with potentially major medicolegal implications. Haematopathologists need to make every effort to remain alert to the possibility of these types of errors and ensure that they are avoided. Figure 1 shows histiocytic necrotising lymphadenitis, a pattern of reactive change with a high risk for the unwary of misdiagnosis as a large cell lymphoma. Other high risks include the misinterpretation of herpesvirus infection, particularly Epstein–Barr virus (EBV), as lymphoma, typically classic HL. Figure 2 illustrates the differential diagnosis of reactive and neoplastic germinal centres. In figure 3, this comparison is extended to include particular reference to the newly recognised category of in-situ follicular lymphoma.13 14 The latter is a particular pitfall as a coincidental occurrence in lymph nodes removed in contexts not raising suspicion of lymphoma, for example, during primary resection or staging of a solid tumour. Another unusual reactive follicular pattern is represented in figure 4, which shows a typical follicle of hyaline-vascular Castleman's disease (HVCD). In addition to recognising that this is not a lymphoma, it is important not to confuse HVCD with the plasma cell type of Castleman's disease (PCCD). Although the latter may be unicentric, polyclonal and innocent it sometimes occurs as multicentric disease with monoclonal plasma cells, usually in HIV-infected patients, requiring different management. In small biopsy specimens such as needle cores, architectural features of PCCD may not be evident (figure 5) and the potential for misdiagnosis of this variant as an extraskeletal plasmacytoma, or as a marginal zone lymphoma with extreme plasma cell differentiation, also needs to be remembered.

Typical example of histiocytic necrotising lymphadenitis, a reactive process that mimics malignancy in lymph nodes. It may occur in the context of Kikuchi's disease (benign, self-limiting, typically involving the head and neck lymph nodes of young women, often of Asian origin) or indicate the presence of underlying autoimmune disease, usually systemic lupus erythematosus. Histopathologists must be familiar with this to avoid misdiagnosis as a high-grade T or B-cell lymphoma due to the abundance of blast cells and activated macrophages associated with high apoptotic activity (insert).

Follicular hyperplasia versus follicular lymphoma. These two follicles from different lymph nodes each show a well-defined mantle zone surrounding a germinal centre. The pallor of the germinal centre in (A) correlates with a high content of centroblasts, which have large nuclei containing only a small amount of dispersed chromatin. The empty holes reflect the presence of scattered tingible body macrophages. Although the germinal centre in (B) may appear less dramatic, its dark colour (comparable with the surrounding mantle zone) is an important clue to its correct interpretation as low-grade follicular lymphoma. The predominant centrocytes are smaller and their nuclei more chromatin-rich than centroblasts, hence the darker and more compact appearance. Proliferative activity is quite different between these two germinal centres. The same germinal centres have been stained for Ki67, a proliferation-associated nuclear antigen, in (C) and (D). The reactive germinal centre (C) shows abundant proliferation, concentrated in one half. This is entirely usual and reflects the zonal organisation of proliferation and selection of cells for apoptosis within the reactive germinal centre. In (D) although neoplastic, the germinal centre of this follicular lymphoma shows very little proliferative activity. Low Ki67 expression within germinal centres should always raise suspicion of low-grade B-cell lymphoma (although not necessarily follicular lymphoma, as other lymphomas may colonise preexisting follicles to produce a similar picture) rather than hyperplasia.
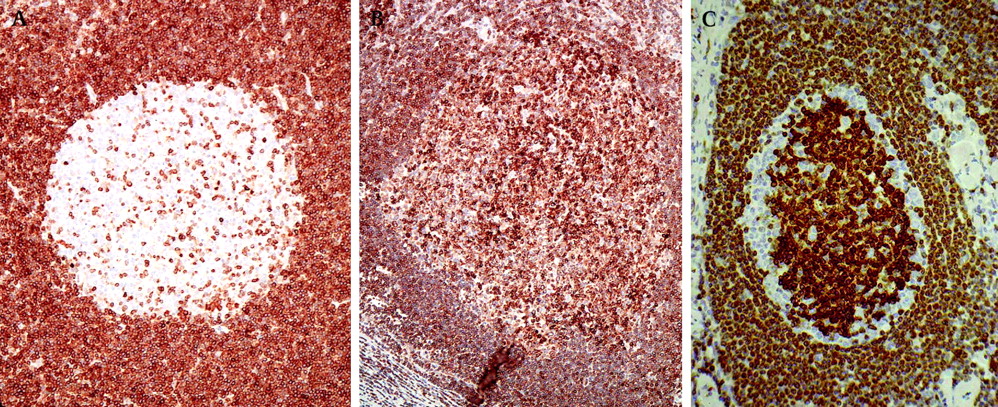
Germinal centre cells in both reactive and neoplastic germinal centres will express CD10, and also BCL6, at least weakly, unless the follicles are colonised by a CD10-negative lymphoma such as mantle cell lymphoma or marginal zone lymphoma. However, reactive and neoplastic germinal centres can usually be distinguished by BCL2 immunostaining. Apoptosis is necessary and abundant in reactive germinal centres, so BCL2 expression is normally downregulated to allow this. Only scattered small T cells are BCL2 positive within reactive germinal centres (A). In most cases of follicular lymphoma, and most other small B-cell lymphomas, BCL2 is expressed by the neoplastic cells. Germinal centres containing BCL2-positive cells in excess of reactive T cells should therefore always be suspected to be neoplastic (B).The pattern shown in (C) represents the recently described in-situ variant of follicular lymphoma. In this variant, nodal architecture is retained and there is subtle colonisation of a proportion of follicles by follicular lymphoma. BCL2 expression is typically particularly strong in such cases, as seen here, and there is usually a variable unstained component of residual non-neoplastic germinal centre cells. The use of the term ‘in situ’ (revised WHO classification, 2008) for this appearance is, however, potentially misleading because this change may be found in multiple lymph nodes and may be accompanied by positive bone marrow staging as further evidence of disseminated lymphoma.

Hyaline-vascular Castleman's disease (HVCD). In this unusual but histologically distinctive reactive condition, follicles appear worryingly dense and dark because of massive expansion of the mantle zone (predominantly containing small, closely packed, B cells). In this condition, mantle zone cells are often arranged in concentric rings around a tiny, pale germinal centre remnant. The latter may contain little more than a small blood vessel and a few scraps of hyaline material (bright pink with H&E staining) representing remnants of a follicular dendritic cell meshwork. The important clue is to recognise that this is a nodule of mantle cells not germinal centre cells, and to then look specifically for the typical highly vascularised tissue in between adjacent nodules of this type. HVCD usually involves a single lymph node or a single group of lymph nodes and is of no sinister clinical significance. It is non-neoplastic and ultimately resolves, although it may persist for a lengthy period. Similar features sometimes affect individual follicles within otherwise ‘usual’ reactive lymph nodes and are not of any known significance in this context.
Plasma cell variant of Castleman's disease (PCCD). This is part of a lymph node needle biopsy core in which the normal cellular mix has been replaced by a diffuse and almost monotonous infiltrate of plasma cells. These were lambda light chain restricted. Without awareness of PCCD, and with only limited tissue to assess in a small biopsy specimen, a mistaken diagnosis of nodal plasmacytoma or even plasma cell myeloma might easily be made. Excision of the complete node, investigation of HIV status and possibly also bone marrow staging are needed to achieve the correct final interpretation.
There are genuine areas of morphological, immunophenotypic and even molecular genetic overlap between HL and NHL15 in rare cases and, more frequently, between Burkitt lymphoma (BL) and aggressive variants of diffuse large B-cell lymphoma (DLBCL).15–18 In these areas, the haematopathologist should discuss the basis of uncertainty with the haematologists/oncologists delivering care for the patient. All clinicians involved need to understand the nature of the uncertainty and ensure that testing (including FISH and other molecular testing if required) is extended as widely as possible to try to achieve a definitive diagnosis. This will involve additional costs, which should be funded without demur in all cases if treatment differs materially between the alternative proposed diagnoses. Ultimately, via multidisciplinary team discussion, a treatment plan should be decided upon mutually, on the basis of an agreed final or ‘best fit’ diagnosis, in the specific circumstances of the individual patient.
Low-grade lymphomas versus high grade
Cell size is a good guide to the grade of many types of NHL. A lymphoma infiltrate composed of relatively monotonous small cells is usually of an indolent, low-grade type and will contain few mitotic figures or apoptotic bodies to indicate high cell turnover. However, failure to appreciate the higher biological grade of some lymphomas composed of small cells can be a cause of misdiagnosis. Mantle cell lymphoma (MCL), lymphoblastic lymphomas and even BL can fall into this category. A particular hazard is the combination of one of these lymphomas with inadequately thin sections. Excessively intense nuclear staining in the latter may mask subtle details of chromatin density and distribution that normally assist in the distinction between mature small lymphocytes, lymphoblastic and Burkitt-type cells. It may also make mitotic and apoptotic bodies difficult to recognise against the tissue background.
Immunostaining for Ki67 can help avoid errors in such diffuse entities, but with nodular or obviously follicular lymphoma infiltrates, Ki67 itself requires interpretation with care, to avoid the mistake of assessing a highly proliferative but still centrocyte-predominant follicular lymphoma, or chronic lymphocytic lymphoma (CLL) with prominent proliferation centres, as more aggressive than the cytological characteristics merit. Individual follicles within cytologically low-grade follicular lymphoma may exhibit high proliferative activity, and a subset of such patients may have more aggressive disease.19 20 However, grading on the basis of proportions of centrocytes and centroblasts is still regarded as the gold standard.21 A high content of proliferation centres in tissue deposits of small lymphocytic lymphoma (SLL)/CLL does not correlate with prolymphocytoid transformation.22 23
HL versus NHL
Distinguishing HL from NHL seems, in principle, to be very easy. Histological criteria for diagnosing HL are generally well known to histopathologists. Indeed, the diagnosis of HL has been regarded as so straightforward by some pathologists that immunostaining has been omitted and no specialist opinion sought. They are partly right; this is not a source of error in most cases, but it represents an example of being right for the wrong reasons. There are genuine biological ‘grey zone’ lymphomas that share features of HL and NHL.15 There are also lymphomas that resemble HL superficially but readily declare themselves to be NHL once immunostained. When no regular immunostaining is used in suspected HL, diagnostic drift is a problem, with failure to recognise atypical cases that might instead be NHL.
Terminology within the current WHO classification is also somewhat unhelpful, because the name ‘nodular lymphocyte predominant Hodgkin lymphoma’ has been retained for a distinct entity that is widely accepted as a variant of B-cell NHL, with a different natural history to that of classical HL.24 Comparison between classic HL and nodular lymphocyte-predominant HL is illustrated in figure 6. Classic HL is characterised by large Reed–Sternberg cells, and variants thereof, scattered in a reactive cellular background. The latter contains a variable mixture of small lymphocytes, macrophages, fibroblasts and other cells such as eosinophils. The presence of candidate mononuclear Hodgkin/Reed–Sternberg cells in lymphoid tissue is insufficient alone to make a diagnosis of HL. Such cells arise in a variety of reactive contexts as well as other lymphomas. Current, highly sensitive immunohistochemistry techniques reveal physiological CD30-positive blast cells in a wide variety of reactive lymphoid infiltrates (figure 7). Large Hodgkin/Reed–Sternberg-like cells are also a frequent finding as a component of some low-grade NHL, most commonly CLL/SLL (often EBV positive in this context), and similar large cells typify large cell lymphomas with anaplastic cytology.

A typical cell mix of classic Hodgkin lymphoma (HL; mixed cellularity subtype) is shown in (A), with Reed–Sternberg cell detail highlighted in the insert. The variant features of nodular sclerosis classic HL are shown in (B) and nodular lymphocyte predominant HL is illustrated for comparison in (C), with ‘L&H’ cell detail highlighted in the insert. Although histological differences are clear in many cases, distinction can be difficult on the basis of morphology alone. The immunophenotype of the large neoplastic cells is, however, quite different in each case: classic HL: CD30+, CD15+*, CD45−, CD20−/weakly +**, CD79a−/weakly +**, BOB1−, OCT2−, EBV+/−, EMA−. NLPHL: CD20+, CD79a+, CD45+, CD30−, CD15−, BOB1+/−, OCT2+/−, EBV−, EMA+/−. The differential diagnosis of these two entities and T-cell-rich large B-cell lymphoma is another difficult and sometimes ‘grey zone’ issue. Currently, the biological, as well as practical, distinction of nodular lymphocyte predominant HL from T-cell-rich large B-cell lymphoma is controversial. *Only a minority of the Hodgkin and Reed–Sternberg cells may be positive in some cases and staining may be subtle—often Golgi-restricted, in approximately 15% of cases, CD15 cannot be demonstrated due to technical limitations arising from the carbohydrate nature of the antigen and the IgM class of reactive monoclonal antibodies. **Typically, both CD20 and CD79a are negative or only one (it can be either) is positive.

Large, CD30-positive immunoblast-like cells in a reactive lymph node. Examined carefully, these rarely approach the size of true Hodgkin or Reed–Sternberg cells and are mononuclear. Additional immunostaining would show CD45 positivity and no CD15 expression. Most of these cells express both CD20 and CD79a, but some are activated T cells, often co-expressing CD25.
The most important mistake of all: DLBCL versus BL
Most errors in haematopathological diagnosis pale into insignificance compared with the importance of correctly identifying BL. Failure to consider high-intensity treatment for appropriate patients with BL, usually reflecting misdiagnosis as DLBCL, may lead to serious undertreatment in patients who might otherwise be cured completely. Cytological variation in DLBCL and BL is wide, with considerable overlap, and morphology alone should never be the basis of distinguishing these entities.
Immunophenotyping is essential and can be very helpful (the neoplastic B cells of BL are typically CD10 positive and BCL2 negative) but variant immunophenotypes are common in BL. The most reliable pointer to a correct diagnosis from standard histology is the Ki67-positive proliferative fraction among the neoplastic cells; in BL this is essentially 100%. However, some DLBCL approach this value and their true biological nature is questioned. Some, but not all, meet current WHO criteria for classification as lymphomas with features intermediate between DLBCL and BL.16 Deregulation of cMYC is fundamental to the pathogenesis of BL and the demonstration of MYC translocation (usually to partner an immunoglobulin heavy or light chain gene locus) by FISH has become crucial in confirming the diagnosis; currently it represents the gold standard. However, this situation is complicated by the occurrence of MYC abnormalities in some DLBCL–NHL, usually associated with additional translocations and/or other numerical or structural chromosomal abnormalities. Such DLBCL are invariably aggressive and their precise relationship to BL represents another current grey zone in lymphoma biology.
Specimen Quality and Diagnostic Accuracy
Box 3 summarises key aspects of specimen quality contributing to correct or incorrect diagnosis and these points are expanded in the following sections.
Specimen limitations* that lead to errors
Small specimen size (eg, needle biopsy and endoscopic specimens)
Crushed or otherwise distorted tissue (eg, mediastinoscopic specimens)
Poor fixation (delay in transfer of fresh tissue into fixative, insufficient fixative volume, insufficient time in fixative, larger specimens not sliced to aid penetration by fixative)
Excessive fixation (formalin pigment deposition obscuring detail, difficult antigen retrieval for immunohistochemistry)
Necrotic tissue
*Note: the importance of developing and maintaining skills in specimen-taking by relevant clinical professionals cannot be overemphasised. Laboratory staff must also pay close attention to ensuring high quality preparation of specimens for histology.
Small specimen size
Endoscopic, punch and needle core biopsy specimens share limitations reflecting, at best, the small amount of tissue that can be obtained. At worst, such small specimens have disproportionate artefacts from trauma (mainly forceps compression) during collection. Small specimens may also be fixed and processed poorly if automated methods are calibrated for larger tissue pieces or if rapid-processing schedules are not optimised. Multiple endoscopic fragments require careful alignment on cellulose strips to ensure adequate representation of each in sections without waste.
Laboratory protocols need to take account of the different requirements from small specimens when lymphoma is suspected, compared with suspicion of a solid tumour or inflammatory pathology. For lymphoma investigation, a single representative and high quality H&E-stained section is the main priority, maximising the preservation of tissue for subsequent preparation of multiple sections for immunostaining, FISH and PCR. For solid tumours and inflammatory processes, H&E-stained sections from multiple levels are usually the major requirements, with a supplementary need to prepare at most a small number of sections from intervening tissue for additional stains. Because not all examples of lymphoma in endoscopic or core biopsy specimens are known or suspected at the time of biopsy, standard laboratory protocols for all such specimens should ensure that sufficient sections are prepared and retained between H&E levels, using appropriately coated or charged slides, for key lymphoma-related immunostains to be performed. A minimum of 10 such sections allows basic immunophenotyping of most common lymphomas. It hardly needs stating that small, precious specimens such as these should not be handled by inexperienced laboratory staff who may not be sufficiently skilled to achieve the maximum use of the tissue.
Poor fixation
Large specimens, such as substantially enlarged lymph nodes and resections of spleen, intestine, lung etc will fix poorly in formalin due to slow penetration of the fixative into bulky tissue. This has detrimental effects on subsequent section quality, the preservation of antigens and the integrity of nucleic acids. This type of problem can largely be avoided if specimens are transferred to the pathology laboratory swiftly after collection from the patient (minutes, not hours) even when placed in formalin before transport. On receipt in the laboratory, such specimens should be examined and sliced without delay to ensure that all or representative areas are exposed to the formalin in slices no more than 10 mm thick.
Crushed and distorted tissue
Mediastinoscopic and needle core biopsy specimens from deep sites are often distorted by marked crush injury, particularly if the tissue is fibrotic. Cellular detail can be rendered uninterpretable as a result and localisation of antigen expression is severely compromised. With needle cores, the narrow diameter of the specimen means that initial sections are usually fully representative unless cut too tentatively in the laboratory. For mediastinoscopic specimens, usually containing multiple fragments, there can be value in proceeding to examine deeper levels. Key immunostains contributory to suspected diagnosis should be undertaken in parallel at each deeper level (or spare sections numbered and stored for use after reviewing H&E-stained slides) to make best use of the tissue. Crush artefacts may be reduced centrally in some fragments, revealed by the deeper sections, and rare features such as Reed–Sternberg cells in HL may be apparent at one level but not another.
Necrotic tissue
Occasionally, a lymph node excised for diagnosis is completely or substantially necrotic, including examples in which necrosis appears to be triggered by previous FNA or needle core biopsy. Little can be done to interpret fully necrotic lymphoid tissues, other than to be aware of the problem and not to over-interpret the histology. The necrosis has become established before the lymph node was removed and no technical magic can re-establish viability. Almost all cellular detail is lost and most immunostains either fail or give misleading, non-specific, positive results. Expression of nuclear antigens is affected early in this process so that Ki67 staining, for example, may seriously underrepresent proliferative activity. Stains for T cells often give false-positive results due to the presence of inflammatory cells in the debris. To a variable extent, CD20 expression is typically retained by B cells and CD79a somewhat more variably so. A reticulin stain may help to demonstrate residual underlying architecture, such as a follicular pattern in follicular lymphoma. Even in the absence of obvious granulomas, the presence of necrosis should always prompt a search for mycobacterial or fungal infection, which may be present as an alternative or additional pathology.
Necrosis occurs in reactive conditions and low-grade lymphomas as well as high-grade lesions. The specific cause is identified only infrequently. Perhaps surprisingly, the relatively minor trauma caused by preceding FNA is sometimes the sole identifiable trigger for necrosis of a lymph node. Necrosis should be anticipated in residual lymph nodes and other masses removed for assessment following chemotherapy for lymphomas (eg, to investigate residual positron emission tomography positivity). In the latter context, examination of sections from multiple levels and selected immunohistochemistry (eg, CD30 in treated HL; figure 8) are frequently needed to ensure adequate scrutiny of the specimen to identify any remaining viable tissue.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lymph node excised due to persistent enlargement and positron emission tomography positivity after treatment for classical Hodgkin lymphoma (HL). Almost all cellular detail is lost (A) within this necrotic mass, enclosed by a rim of dense reactive fibrous tissue. There is unsatisfactory, equivocally positive CD30 immunostaining in parts of the necrotic tissue (B). Only examination of the small peripheral areas of viable lymph node revealed tissue containing a cell mix consistent with HL (C), with reliably positive CD30 immunostaining (D).
Errors relating to immunohistochemistry
Box 4 summarises the main sources of such errors, and some of these are discussed in more detail in the following sections.
Errors relating to immunohistochemistry
Insufficient understanding of antibody reactivities
Use of inappropriate positive and negative controls
Sensitivity changes due to alterations in reagents or machines used
Insufficient range of tests used, due to cost concerns
Drift in performance of reagents if use insufficient to ensure adequate turnover and replacement
Understanding antibody reactivities
This is a large topic that cannot be covered in detail here. The most important principle is to understand that a monoclonal antibody has specificity for one epitope of one antigen and not, per se, for the cell expressing that antigen. Therefore, such an antibody may be lineage associated (eg, CD20 and B cells; CD3 and T cells) but is not lineage specific. The CD (cluster of differentiation) numbers assigned to antigens and their corresponding monoclonal antibodies were introduced partly to clarify this relationship. However, the structure of some antigens is complex, giving rise to multiple epitopes. Consequently, some antibody clones (eg, OPD4 and UCHL1 directed against CD45RO) have overlapping but different spectra of reactivity in immunohistochemistry (OPD4 reacts less than does UCHL1 with CD45RO expressed by myelomonocytic cells). In some cases, the spectra are sufficiently divergent that the CD numbering has been adjusted to recognise the variation; for example, CD68 (expressed by granulocytic and monocytic cells and reactive with antibody clone KP1) and CD68R (more selectively expressed by monocytic cells and reactive with both KP1 and PGM1). If these overlaps and differences are not understood, errors in interpretation may arise.
It is also crucial to understand the normal distribution of antigens and their anticipated variation in neoplasia. This applies equally to the consideration of immunohistochemistry in the diagnosis of solid malignancies, such as carcinomas, sarcomas and malignant melanomas, but lymphoid and lymph node accessory cells are the main concern in this review. A key example in which misunderstanding occurs regularly is the value of detecting BCL2 expression in the context of suspected follicular lymphoma. There is no BCL2 expression by reactive germinal centre B cells but, as a result of the translocation t(14;18) in follicular lymphoma, this antigen is expressed inappropriately in the latter. However, expression is also normal in most T lymphocytes and so reactive germinal centres rich in the latter will contain abundant positive small cells; it is important to look closely for expression specifically by germinal centre B cells and to compare with the expression pattern of a strongly T-cell-associated antigen such as CD3. It is also important to recognise that most peripheral B cells, apart from those within physiological germinal centres, are normally BCL2 positive and so are the neoplasms derived from them (MCL, marginal zone, lymphoplasmacytic and lymphocytic lymphomas). Consequently, BCL2 expression has no differential diagnostic value when attempting to distinguish between these entities. At the very least, the use of this immunostain in such a context is a waste of money; at worst, it could lead to an erroneous diagnosis if BCL2 expression if mistakenly believed to be limited to follicular lymphoma. The WHO classification monograph,25 while not entirely comprehensive in its coverage of immunohistochemistry in lymphoma diagnosis, contains a valuable summary of the anticipated immunophenotype for each of the entities described and is a key source of reference.
A further source of misunderstanding is a failure to recognise the non-equivalence of reactivities/techniques; an important example here is the detection of EBV by immunostaining for latent membrane protein-1 (LMP1) versus detection by in-situ hybridisation for Epstein-Barr virus early RNA (EBER). Detection of LMP1 is convenient and suitable for the demonstration of EBV in the latency state associated with HL but should be avoided in almost all other contexts in which EBV is suspected as the underlying driver of lymphoid cell proliferation. It is not expressed during productive viral infection, as in infectious mononucleosis, or in many latency states associated with large B-cell proliferations, including those overtly associated with immune compromise (post-transplant lymphoproliferative diseases, HIV-associated lymphomas, etc). In contrast, EBER are expressed in all of these contexts, including HL-associated latency. Their detection by in-situ hybridisation is preferable overall if a single technique is to be adopted by an individual laboratory; methods for EBV–EBER in-situ hybridisation have become increasingly harmonised with standard immunohistochemistry in recent years, including suitability for automation. The danger of relying on LMP1 expression is the occurrence of false-negative results in many lymphoid proliferations other than HL.
Other examples relate to the investigation of common translocations in B-cell lymphomas, such as t(11;14) and t(14;18), which may be detectable by FISH in the absence of protein overexpression (cyclin D1 and BCL2 in these examples) at a level detectable by immunostaining. Better appreciated is the lack of direct concordance between the results achievable using flow cytometry for immunophenotyping and those from immunohistochemistry. Not all of the variation, however, is attributable to the deleterious effects of tissue fixation on antigen expression, which is a common assumption. Different antibody clones have been optimised for use in one or other of these techniques and do not necessarily have absolutely identical reactivities; examples would be the differential detection of CD34 and terminal deoxynucleotidyl transferase (TdT) epitopes by the different clones commonly used in these two techniques.
Immunostaining with polyclonal antisera, rather than monoclonal antibodies, is generally employed to demonstrate light and heavy chains (except delta and epsilon chains, for which good monoclonal reagents are available). Non-specific background staining is common when using polyclonal antisera, a problem that is exaggerated by excessive proteolysis for antigen retrieval. In addition, most tissues contain abundant immunoglobulin in plasma, lymph and tissue fluid, giving further unwanted staining. High background is particularly a problem with commonly used anti-kappa and anti-mu antisera; there is therefore a danger of either over or underinterpreting these immunoglobulin light and heavy chain stains when overall staining of tissue is intense. Depending on the technical skills available, do not expect too much from immunohistochemistry for immunoglobulins.
For kappa and lambda light chain demonstration in plasma cells, in-situ hybridisation with messenger RNA offers a simple and, in most laboratoriess, greatly superior technique. Simultaneous dual-colour detection of both light chain mRNA types is also possible and is not technically difficult. Unfortunately, the sensitivity of current in-situ hybridisation methods is insufficient to detect reliably the tiny amounts of light chain mRNA in other B-lineage cells and immunostaining remains more informative in lymphomas.
Inappropriate controls may mislead
The fundamental principle underpinning the selection of appropriate controls for immunohistochemistry is that relevant, rather than simply convenient, tissue must be used. Maintaining supplies of relevant positive control tissue can be difficult, but it is essential.
It is also essential to know what internal control reactivities to expect in normal/reactive as well as neoplastic lymphoid tissues. Internal controls are elements of the diagnostic tissue itself that help provide reassurance that immunostaining has worked. Knowledge of these is gained predominantly by experience, and normal reactivities should not be misinterpreted as pathological; for example, endothelial cell expression of CD34 and expression of cyclin D1 by occasional bone marrow stromal cells.
Traditionally, reactive tonsil has provided the standard positive control tissue for immunohistochemistry relating to lymphoid cells. With an expanding repertoire of antibodies available, it remains very useful in many, but not all, circumstances. For example, although CD5-positive T cells will be abundant in tonsil, validation of staining of CD5-positive neoplastic B cells in the differential diagnosis of B-cell lymphomas requires the use of positive lymphoma tissue (such as SLL or MCL). Likewise, although CD15 and CD30 are expressed by neutrophil polymorphs (CD15) and reactive immunoblastic lymphoid cells (CD30) in tonsil, HL tissue with neoplastic cells known to be positive for each antigen should be used when investigating suspected HL. For anaplastic lymphoma kinase (ALK; CD246) expression, only in ALK-positive large cell lymphoma is suitable as positive control tissue.
In developing CD10 monoclonal antibodies, commercial suppliers have taken advantage of strong expression of this antigen by some epithelial cell types, particularly renal tubular epithelium, and some recommend the use of kidney for positive control tissue. This is clearly inappropriate in haematopathology—in this case, reactive tonsil is an appropriate alternative, as long as germinal centres are prominent.
Immunohistochemistry: variation in sensitivity
Most monoclonal antibodies in common use as diagnostic reagents have been optimised for performance with fixed, paraffin-embedded tissues and modern automated immunostaining systems. However, some still stain with less sensitivity than would be ideal, varying with fixation, so that true positive lymphoma cases may be missed; CD5 is an example in this category.
Some antibodies stain different cells within pathological tissue with varying intensity, and it can be difficult to decide where to draw the line between positive and negative. This represents a genuine biological ‘grey scale’ of antigen expression and can confound the accurate assessment of immunostains for antigens such as Ki67, p21 and p53.
Other antibodies are oversensitive as currently used and react with physiological as well as neoplastic cells. A false-positive diagnosis may be made if the former are not recognised as such. An example of this type is CD30 detection in reactive immunoblasts (as illustrated in figure 7) in addition to neoplastic large cells of HL and anaplastic large cell lymphoma.
These problems apply to staining in all immunohistochemistry laboratories but the precise thresholds at which potential difficulties may arise will differ. They will also change over time as reagents and automated instruments evolve. Detailed familiarity with the particular output of your local laboratory is essential.
Errors arising because too few tests are done
At the simplest level, with an insufficient range of tests performed there may be inadequate diagnostic precision; for example, leaving a diffuse small lymphoid cell infiltrate categorised as small B-cell lymphoma without investigating CD5, CD23, cyclin D1 and Ki67 expression to establish a definitive diagnosis (CLL, MCL, marginal zone lymphoma, etc). Even with stains sufficient to establish the diagnosis, there is an increasing need to evaluate additional immunohistochemistry for prognostic assessment; for example, CD10, BCL2, BCL6 and IRF4/MUM1 in DLBCL to determine the germinal centre or non-germinal centre type.26 Several additional algorithms involving antibody combinations based on gene expression profiling have been proposed for improved prognostication in aggressive B-cell lymphomas. We can anticipate that the required range of testing will expand.27–29 Scoring antigen expression meaningfully for use in such algorithms needs careful attention to cut-off points between positive and negative staining30 and the development of consensus criteria for assessment may be crucial.
The use of an insufficient range of immunostains may reflect limitations of local availability (a major factor underpinning the ongoing moves to centralise haematopathology diagnostics in large regional centres) or failure to appreciate the full differential diagnosis in individual cases. Examples in which an insufficiently wide differential diagnosis may be considered are shown in box 5.
Errors arising from consideration of insufficiently wide differential diagnosis: some examples
Misleading range of investigations undertaken:
High-grade plasmablastic tumours misinterpreted as non-haemopoietic because of downregulation of lymphoid cell-associated antigens
Extramedullary presentations of acute myeloid leukaemias mistakenly interpreted as aggressive lymphoma in the absence of screening for myeloid differentiation
Neoplastic mast cell infiltrates interpreted as histiocytic, supported by CD68 expression but without concurrent staining for tryptase and/or CD117 to establish correct phenotype
Opportunity to find subtle pathology by screening missed:
Missed ‘in situ’ follicular lymphoma in reactive-appearing germinal centres
Opportunities to maximise diagnostic/prognostic precision missed:
Peripheral T-cell lymphoma, not otherwise specified, diagnosed instead of lymphomatous adult T-cell lymphoma/leukaemia, if CD25 not included in test panels
DLBCL or peripheral T-cell lymphoma, not otherwise specified, diagnosed instead of distinctive B and T-cell lymphoma subtypes associated with EBV expression, if EBV–EBER in-situ hybridisation not included in test panels
Errors relating to molecular genetic tests
A limited range of molecular tests has gained an established role in haematopathology in recent years. These include clonality assessment by analyses of IGH, IGK, IGL, TCR-B and TCR-G rearrangements by PCR31–33 and a limited range of FISH translocation analyses (eg, IGH-BCL2, CCND1-IGH, BCL6, MALT1 and MYC rearrangements).34 35 When undertaken using fixed tissue with suspected lymphoma, PCR results must always be interpreted with reference to both the quality of the DNA template, influencing the technical quality of the PCR, and the histological context, including immunophenotype. False-negative results may arise from insufficient quantity or inadequate quality of DNA, or from the use of primer sets too limited in scope to provide high sensitivity. The latter has been minimised by programmes such as the European Union Biomed-2 project, which have provided harmonised, comprehensive IGH, IGK, IGL and TCR primer sequences optimised for use in multiplexed PCR and with fixed, paraffin-embedded tissues.31–33 False-positive results may arise through PCR contamination, which should not occur with good laboratory practice, or failure to appreciate that a clonal result does not necessarily equate with clinically neoplastic behaviour by lymphoid cells. A ‘pseudo-clone’ may be pre-neoplastic or a forme fruste of neoplasia or may occur in the highly focused physiological immune response to a super-antigen.
New PCR-based molecular tests are likely to become important in the next few years, such as micro-RNA expression36 and the assessment of target gene hypermethylation.37 These will take us progressively further away from traditional reliance on morphology in lymphoma assessment and will require new knowledge by molecular scientists and haematopathologists for appropriate interpretation.
The technical and interpretational challenges of FISH vary greatly depending on the cellular substrate (FNA, fresh tissue imprint, extracted fixed nuclei, whole fixed tissue section). The appropriate use of both co-location and break-apart probes is essential to avoid false-negative results in which translocation partners may vary. Most haematopathologists are aware of their limited expertise in interpreting FISH undertaken using tissue sections, which is an emerging skill among cytogenetic and molecular genetic laboratory staff. It can be very helpful to provide a relevant immunostained section, with interpretation/annotation in complex cases, to aid the selection of nuclei for assessment in the laboratory. It remains crucial to be aware of the potential for false-negative results as a result of nuclear truncation or poor fixation. As for PCR, FISH results require careful interpretation, best achieved by collaboration between pathologists and laboratory scientists and always with due attention to histological context.34 35
Finally in this section, because PCR and FISH studies are generally undertaken as additional analyses after histology and the immunophenotype have been assessed and reported, it is important to avoid errors or omissions in communication that may arise as a result of prolonged delay, or complete failure, in providing additional reports. This requires good organisational skills and its importance should not be underestimated.
Conclusions
Avoiding errors in the diagnosis of lymphomas and related conditions requires a considerable breadth and depth of knowledge,38–40 but also other skills of equal importance. Confidence to request a range of investigations from laboratories outside the cellular pathology department is needed. A willingness to engage in dialogue with a variety of clinicians and scientists is essential to maximise the information shared and the appropriateness of investigations and their interpretation. With such a complex set of requirements, excellent organisational skills are also vital to ensure that complete, integrated and timely reports are produced.
To finish, one further type of error should be re-emphasised. It is possible to make a correct diagnosis by (happy) accident. Of course, this is not truly accidental and its occurrence is certainly not restricted to haematopathology. It occurs when the apparent familiarity of a particular, often common, pathological pattern is not challenged, through lack of awareness of alternative, maybe rare, diagnoses. This is the bear-trap awaiting any wallpaper-matchers in the world of histopathology. In haematopathology, reluctance to undertake immunostaining in ‘obvious’ HL or ‘barn-door’ follicular lymphoma is a reflection of this type of thinking. Pathologists taking such risks deserve their fate—the accident will not be so happy on another occasion!
Take-home messages
The correct reporting of lymph node specimens requires the integration of a wide range of clinical, laboratory and imaging information; histology should not be assessed in isolation. Key components of this integrated approach to lymph node interpretation are:
Adequate and well fixed specimens, ideally with the opportunity for immediate sampling before fixation to undertake investigations requiring fresh cells or frozen tissue.
Access to detailed background clinical information and results of additional tests (in particular, haematology investigations and radiological imaging).
Recognition that there are very few circumstances in which an H&E-stained section is sufficient to assess a lymph node removed for suspected haematological malignancy.
Use of preselected antibody panels for immunostaining and familiarity with the expected results, including controls.
Increasingly, experience in interpreting additional molecular studies such as clonality PCR and FISH.
Familiarity with the major categories of the WHO lymphoma classification and their differential diagnosis.
Awareness of ‘high-risk’ differential diagnoses (eg, reactive vs neoplastic; high vs low-grade lymphoma) including robust strategies to reach the correct diagnosis in such cases.
A collaborative approach to working with diverse clinical and laboratory colleagues, plus excellent report-writing skills.
References
Footnotes
Competing interests None.
Provenance and peer review Commissioned; not externally peer reviewed.