Article Text

Abstract
Barrett's oesophagus is important as a precursor of oesophageal adenocarcinoma via a metaplasia-dysplasia-carcinoma sequence. It is often detected on upper gastrointestinal endoscopy. In the absence of glandular dysplasia the risk of progression to cancer is low but ascertainment of dysplasia is not always straightforward. Sparse mucosal sampling may miss dysplasia, or reactive changes may be overinterpreted due to inter and intraobserver variation. Low-grade and even high-grade dysplasia do not necessarily progress, provided prevalent cancer has been rigorously excluded. This indeterminacy motivates an ongoing search for clinically useful predictive biomarkers. Although many genetic and epigenetic abnormalities have been associated with neoplastic progression in Barrett's mucosa no molecular tests have as yet been accepted into routine pathology practice. Challenges of assay definition remain and many marker studies lack statistical power or have other methodological flaws. Even where strong evidence of clinically relevant predictive value does exist (in the case of ploidy analysis by flow or image cytometry) adoption has been minimal, likely reflecting technological and possible reimbursement obstacles. Well designed multicentre studies are likely to be required to translate improved knowledge of Barrett's carcinogenesis into clinically significant progress on predictive testing, and will require a degree of cooperation not so far widely seen in the field.
- Barrett's oesophagus
- biological markers
- risk assessment
- preneoplastic conditions
- histopathology
- diagnostics
- malignant tumours
- oesophagus
- breast
- morphometry
- 3-D reconstruction
- stomach
Statistics from Altmetric.com
- Barrett's oesophagus
- biological markers
- risk assessment
- preneoplastic conditions
- histopathology
- diagnostics
- malignant tumours
- oesophagus
- breast
- morphometry
- 3-D reconstruction
- stomach
Introduction
Barrett's oesophagus and oesophageal adenocarcinoma
Oesophageal adenocarcinoma has increased fivefold in the UK and the US over the past two decades, while the incidence of oesophageal squamous cancer has been static.1 Despite improved preoperative staging and perioperative care, most oesophageal cancers remain advanced at diagnosis, with 5 year survival around 10% to 20%.2
Barrett's oesophagus is an acquired precursor of oesophageal adenocarcinoma (figure 1), and the incidence of both conditions has risen in parallel.3 However oesophageal adenocarcinoma may develop in the absence of Barrett's oesophagus.3 Characteristically, the squamous epithelium of the lower oesophagus is replaced by a metaplastic glandular mucosa.4 The British Society of Gastroenterology definition is ‘an endoscopically apparent area above the oesophagogastric junction that is suggestive of Barrett's (mucosa) which is supported by the finding of columnar lined oesophagus on histology’. This definition does not demand intestinal metaplasia (IM) with goblet cells to be present,5 unlike definitions current in the USA and Europe (but not Japan), which do.6 7

Invasive adenocarcinoma 15 mm in diameter (box) arising in a C3M6 Barrett's oesophagus (Prague criteria in which C3 describes 3 cm of circumferential Barrett's epithelium and M6 is the total length of the metaplastic segment, in this case 6 cm). Despite its relatively small size this lesion has invaded muscularis propria (pT2) and metastasised to two regional nodes (pN1). It has developed on a background of extensive low-grade and high-grade Barrett's dysplasia. This is a late stage in the metaplasia-dysplasia-carcinoma sequence in a woman patient, relatively young to have developed a Barrett's adenocarcinoma (age 48).
It appears that reflux of gastric and duodenal contents leads to mucosal injury, cellular proliferation and columnar/glandular metaplasia of the normal squamous mucosal lining, although the details are not fully elucidated.8 A metaplasia-dysplasia-cancer sequence is characteristic of progression to Barrett's adenocarcinoma.9 Barrett's oesophagus is common in the Western world, especially in people with chronic reflux. About one-third of Western adults experience heartburn at least once a month, one-third of whom will have endoscopic oesophagitis. Of these 40% will improve spontaneously, 50% will have persistent symptoms and 10% will progress to Barrett's oesophagus. Patients with Barrett's oesophagus are about 30 times more likely to develop oesophageal adenocarcinoma than patients without it, but even so only 0.5% to 1% will progress to adenocarcinoma annually with a lifetime risk of 5% in men and 3% in women with Barrett's oesophagus.1 4
Current guidelines on both sides of the Atlantic suggest that patients with Barrett's oesophagus should undergo periodical surveillance endoscopy with biopsy. In those without dysplasia endoscopy is recommended every 2 years and in those with low-grade dysplasia, every 6 months, although evidence of surveillance effectiveness is sparse. The treatment of high-grade dysplasia is complex, with options including frequent endoscopic surveillance and comprehensive biopsy, and endoscopic therapies including mucosal resection, argon plasma coagulation and radiofrequency ablation, with oesophagectomy reserved for cases not treated by other means.
Recent UK National Institute of Health and Clinical Excellence guidelines recommend that patients with high-grade dysplasia or intramucosal carcinoma should be discussed in an relevant multidisciplinary forum and offered endoscopic mucosal ablation if appropriate.10 At present, it is neither feasible nor cost effective to endoscope all patients with reflux symptoms. As most patients with Barrett's oesophagus never develop an adenocarcinoma, most would derive no benefit and some might even be harmed by expensive and relatively invasive endoscopic and biopsy surveillance.11 12
The potential for biomarkers
In this context, a clinical or laboratory marker which did actually predict progression to dysplasia (itself a marker of cancer risk), or to cancer in the case of patients with dysplasia already, would be extremely valuable, by allowing targeting of screening to those most at risk. Currently, high-grade dysplasia is the most reliable indicator of increased risk of progression to malignancy, and indeed it is often already associated with invasive cancer when detected.13 14
However, estimates of the incidence of progression from dysplasia to carcinoma are variable, and the diagnosis of dysplasia, particularly low-grade dysplasia, can be difficult owing to sampling errors, disagreement between observers and the difficulty of discriminating inflammatory and reactive changes from true dysplasia.13 Despite a substantial literature on genetic and molecular changes in Barrett's oesophagus and Barrett's adenocarcinoma, practically nothing has translated so far into clinical practice. The aim of this review was to examine candidate molecular and other biomarkers in Barrett's oesophagus, and prospects for progress.
Literature search
A search of the English language literature since 1975 was performed using PubMed, Embase and Cochrane databases with MeSH terms ‘Barrett's oesophagus/epithelium/metaplasia’, ‘oesophageal cancer/adenocarcinoma’, ‘biomarkers’, ‘disease progression’ and ‘dysplasia’. Abstracts were examined and relevant articles from reference lists of other papers retrieved.
Biomarkers
Biomarkers should detect a state already established, predict a future state, or both. Few achieve either. Good biomarkers require high sensitivity and specificity for the state or event they purport to detect: cardiac troponins, for example, are excellent markers of myocardial injury but are not highly specific for myocardial infarction. In Barrett's oesophagus we might wish for markers of the diagnosis per se; but especially useful would be predictors of premalignant or malignant progression. A priori, such markers are likely to be concerned with key pathways in the development of oesophageal adenocarcinoma and, to be useful, would distinguish clearly between people with low and high cancer risk. Ideally a test would be minimally invasive, cost effective and could be used on its own or in conjunction with other techniques.15
A formal model of phased biomarker development has been proposed, analogous to the process in therapeutic drug studies.16 These are: (a) preclinical exploratory studies to identify potential markers; (b) clinical assay development to determine sensitivity and specificity of markers in subjects with the disease, compared to normal control subjects; (c) retrospective studies on specimens from subjects prior to their diagnosis, to test capacity of the marker to detect preclinical disease; (d) prospective screening studies; and (e) cancer control studies to address whether screening with biomarkers reduces the population burden of cancer.
Biomarkers never studied beyond phases 1 or 2 vastly outnumber those taken to phases 3 and 4, and there are few established, clinically useful predictive biomarkers in Barrett's oesophagus, other than histomorphology. Even so, there are markers for which evidence of predictive power does exist, but which are used only in a few centres, in particular ploidy, assessed by flow or image cytometry. In ploidy studies relatively complex technology and reimbursement issues may have impeded wider adoption.
Some believe panels of biomarkers may eventually provide more useful clinical information than any single marker, but the validation challenges for marker combinations will be at least as great as for single markers.
To evaluate the prospects for new biomarkers, we need to understand and define morphological molecular and genetic abnormalities associated with Barrett's oesophagus.
Morphological features of Barrett's oesophagus
Intestinal metaplasia
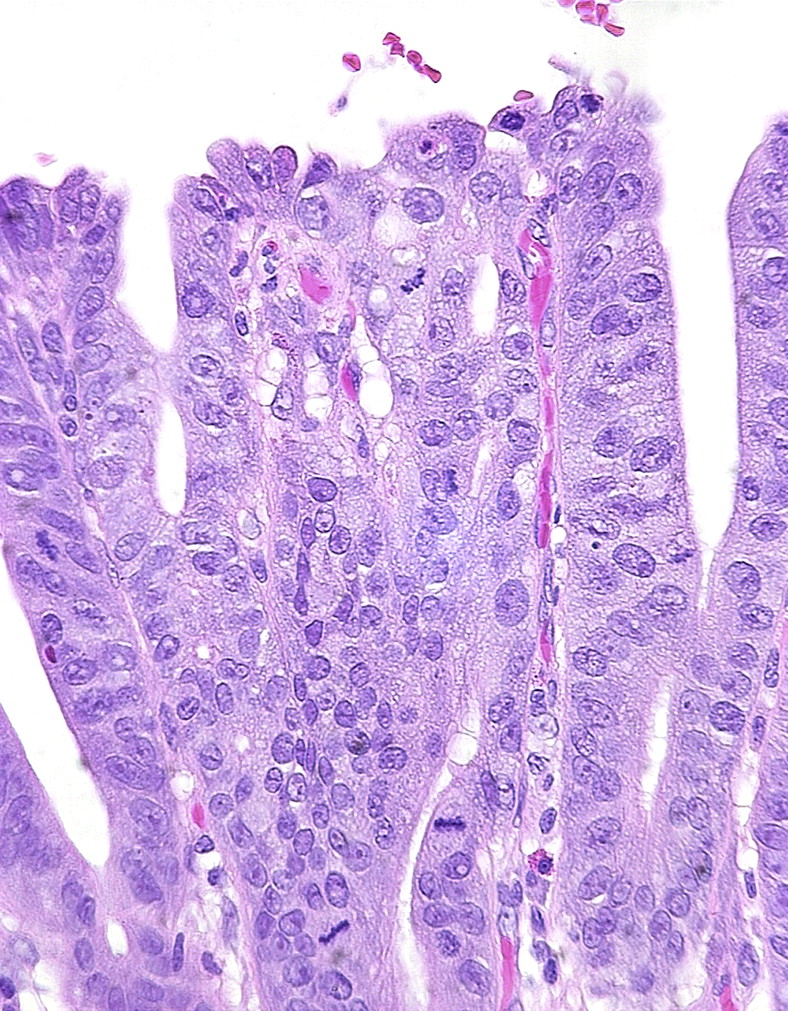
IM per se (figures 2 and 3A) is not a useful marker of cancer risk, being present in most cases if not every case of Barrett's oesophagus, but the belief that oesophageal glandular dysplasia and adenocarcinoma usually develop on a background of IM has led to a perception that without IM cancer risk may be low. Recent data suggests this is an oversimplification.17 Studies suggest that a columnar metaplasia of the oesophagus carries the same malignant risk whether an intestinal phenotype is present or not, and therefore the original association of IM carrying malignant risk has been brought under scrutiny. Also, even in the absence of detectable goblet cells, Barrett's mucosa still expresses markers and has ultrastructural features of intestinal differentiation (figure 3B). The perception that an absence of goblet cells negates a diagnosis of Barrett's oesophagus may therefore not survive18 but will not be considered further here.

Characteristic diversity of mucosal phenotypes in Barrett's oesophagus. A. Cardiac-like crypt without any goblet cells. B. Point (*) on mucosal surface where cells from crypt in (A) meet cells from crypt in (C). C. Neighbouring intestinal-type crypt with numerous goblet cells. There is no dysplasia in any of these fields.
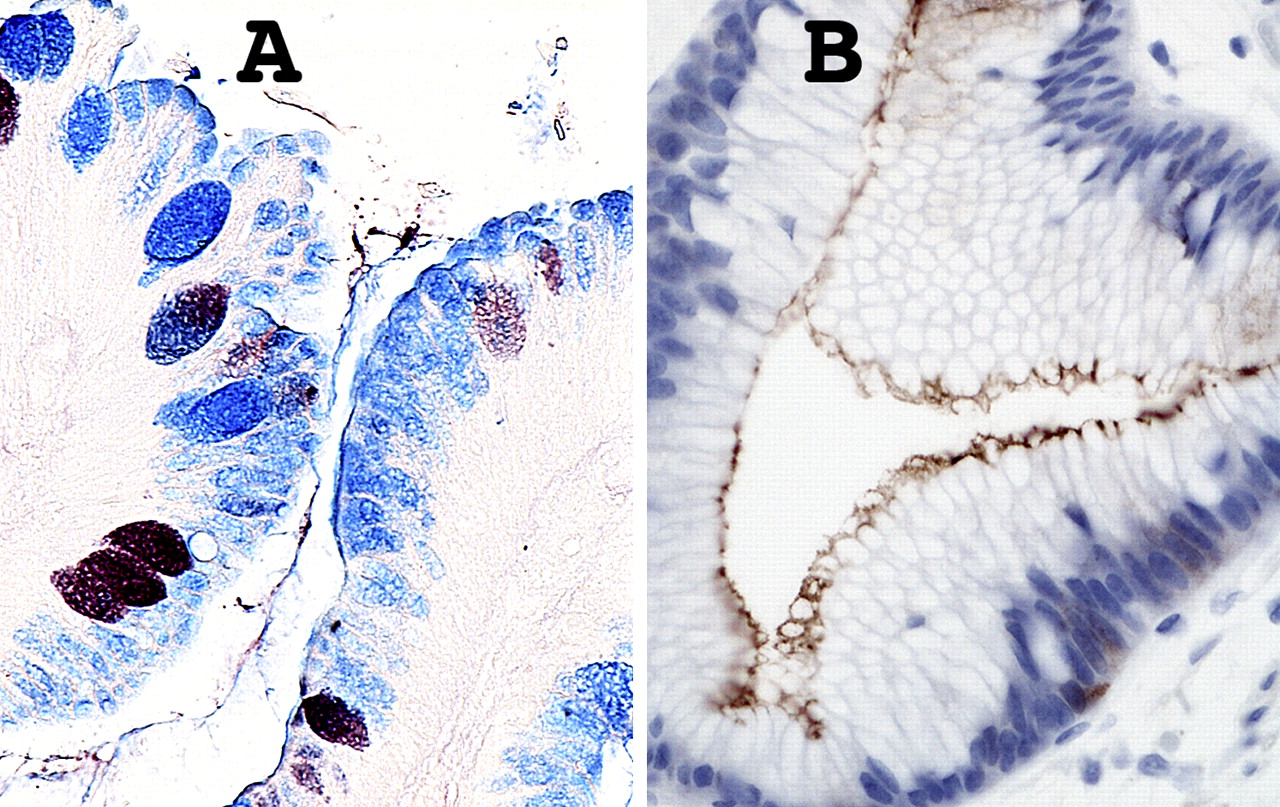
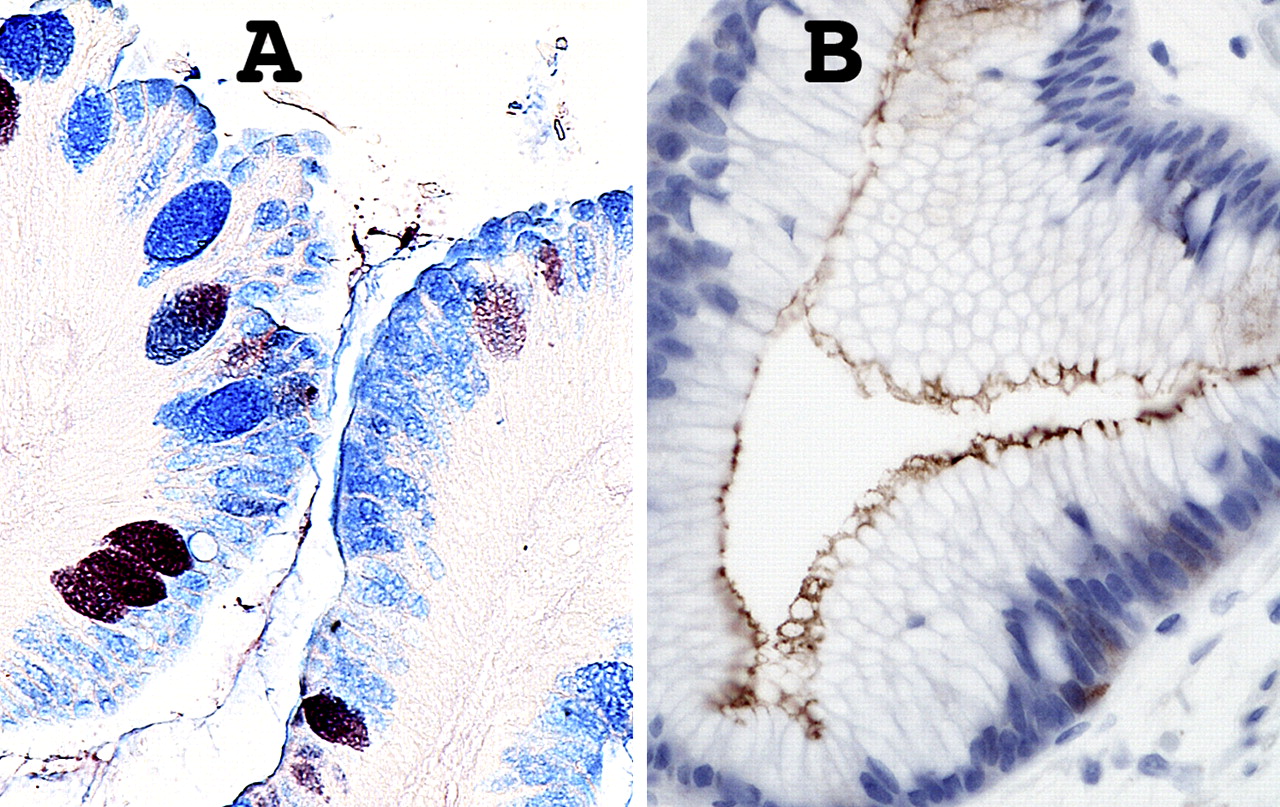
Further mucosal phenotypic diversity in Barrett's oesophagus. A. Alcian blue pH 2.5 and high iron diamine staining of sialylated and sulfated acidic mucopolysaccharides. Goblet cells may contain sialylated (blue), sulfated (brownish-black) or mixed mucopolysaccharides. Intervening columnar cells stain blue, implying that they contain sialylated acidic mucopolysaccharides. Such ‘columnar blue’ cells may be present with or without goblet cells. In the absence of goblet cells ‘columnar blue’ cells are inconsistent with a purely gastric mucosal phenotype. B. Barrett's mucosa with a cardiac-like phenotype. There are no goblet cells, but immunostaining for the intestinal marker villin is clearly positive along the apical border of the columnar cells, implying intestinal differentiation even in the absence of goblet cells.
Dysplasia
As in other situations dysplasia is a marker of cancer risk in Barrett's oesophagus. In the gastrointestinal tract it is synonymous with intraepithelial neoplasia, and implies architectural and cytological changes commonly associated with carcinomas, and from which the latter are presumed, at least sometimes, to have evolved. As a marker of risk, however, dysplasia is far from perfect. There is significant intra and interobserver variation in assessing Barrett's oesophagus.13 Pathologists are not good at agreeing on the presence of mild and moderate (low-grade) dysplasia (figure 4), although agreement on severe (high-grade) dysplasia (figure 5) is better.13 19 Dysplasia may be patchy, and many biopsies may be necessary to detect it reliably, creating a burden for patient, endoscopist and pathologist.20 Not all dysplasia will progress to higher-grade or invasive adenocarcinoma (figure 6); in some cases it may regress.

Morphologically heterogeneous Barrett's mucosa including low-grade dysplasia. Same patient as figure 1. At * there is no dysplasia but at ** there is cellular stratification and proliferation involving the mucosal surface. A tripolar mitosis (inset) strongly supports the diagnosis of dysplasia.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Well differentiated Barrett's adenocarcinoma. Same patient as figures 1, 4 and 5. Irregular glands and occasional individual carcinoma cells are embedded in an oedematous/desmoplastic stroma. This field was intramucosal in location but similar invasive carcinoma was present in submucosa, invading muscularis propria and in local lymph nodes. The stromal reaction is not always so well developed, even in lesions proven to be invasive by having undergone metastasis.
Even high-grade dysplasia, provided invasive adenocarcinoma is not already present, may persist for years before progression to invasion. All of these considerations emphasise the limitations of dysplasia as a risk biomarker. That no better biomarker has yet emerged is only to restate the challenge. It is likely that dysplasia will remain a mainstay of risk assessment in Barrett's oesophagus for some time, with newer technologies complementing it initially, not least because morphology may usefully allow targeting of marker studies such as ploidy assessment by image cytometry, immunohistochemistry, or FISH (fluorescent in situ hybridisation). Different patterns of dysplasia are also coming to be recognised and in time may prove to have different behaviours.21 22 Until then, dysplasia agreed on by more than one experienced gastrointestinal (GI) pathologist may be more robust than an uncorroborated diagnosis. Where three pathologists agree on a diagnosis of low-grade dysplasia, an elevated risk of progression exists,23 perhaps because dysplasia on which any three GI pathologists can agree is close to being high grade.
Molecular abnormalities of Barrett's oesophagus
Mutations accumulating in premalignant tissue lead to evolution of cellular clones with increasing genomic instability and abnormal cell behaviour until clones of cells emerge with invasive and metastatic potential.24 25 Epigenetic events and aspects of the host environment such as inflammation are also important, and cancer can be promoted by factors not known to be genotoxic.
Genomic instability is a fundamental property of neoplastic progression, developing before the onset of cancer and characterised by chromosomal instability (aneuploidy), epigenetic instability, loss of heterozygosity (LOH) affecting tumour suppressor genes and microsatellite instability. The targets of genomic instability are usually seen to include proto-oncogenes, tumour suppressor genes, DNA mismatch repair genes and mitotic checkpoint genes.26
This classification has limitations: some newer markers cannot easily be classified in these groups. Hanahan and Weinberg's popular taxonomy of properties required by cancer cells (namely, growth self-sufficiency, insensitivity to growth inhibitory signals, avoidance of apoptosis, replication without limit, sustained angiogenesis, invasion and metastasis) provides a convenient framework for an examination of potential biomarkers.27 28 However over the last decade the cancer stem cell hypothesis has caused a shift in thinking about the key events of carcinogenesis. Stem cells and cancer cells share several important properties and there is now evidence to suggest dysregulation of the self-renewal process of stem cells may be the key event in early carcinogenesis rather than random mutation. This hypothesis, if adopted, carries significant implications for diagnosis and therapeutic options in cancer.29
Growth self-sufficiency
Normal cells require exogenous growth signals to move from G0 (quiescence) into the cell cycle. A key mechanism controlled by the retinoblastoma protein p185 Rb, late in G1, restricts progression into S phase and DNA synthesis to cells without DNA damage, which may trigger cell cycle arrest and DNA repair, or if the damage cannot be repaired, apoptosis and cell death.30
Cyclins and cyclin dependent kinases
Progression through the cell cycle is controlled by cyclins and cyclin dependent kinases (Cdks). Different Cdks and cyclins are required at various stages of the cell cycle. There are two main structurally related groups of Cdk inhibitors. The Ink4 family (‘inhibitors of Cdk4’) consists of proteins (p15, p16, p18, p19) that inhibit cyclin D-Cdk 4/6 complexes. Mutations, deletions or silencing through DNA methylation of p15 and p16 have been reported in various human malignancies. The other group is the Cip/Kip family including p21, p27 and p57, which preferentially target Cdk2. p21 (also known as Cip1 or Waf1) is regulated by p53, and although there are many mutations in p53, no molecular alterations of p21 have yet been reported.
Cyclin D1
Cyclin D1 is a proto-oncogene that controls the G1-S transition by activating Cdks 4 and 6, which phosphorylate p185 Rb (thereby inactivating it) and stimulate progression through the cell cycle. A single base polymorphism in a variant known as cyclin D1b has been implicated in overexpression and neoplastic transformation, and immunohistochemistry shows cyclin D1 overexpression in Barrett's oesophagus and oesophageal adenocarcinoma.31 It has been claimed that patients with Barrett's metaplasia and cyclin D1 overexpression are 6–7 times more likely to develop adenocarcinoma32 but other studies do not support this33 and while increased expression of cyclin D1 is an early event in adenocarcinogenesis and may of itself predispose to malignant transformation,34 at present abnormalities of cyclin expression cannot be confirmed as markers of progression risk.
Cyclin A is also a proto-oncogene expressed in the proliferative compartment in normal gastrointestinal mucosae. Cyclin A immunohistochemistry from oesophageal brushings in patients with Barrett's showed similar localisation in the proliferative compartment in 76% of samples. However with increasing grades of dysplasia, the expression of cyclin A shifted toward the upper crypts and surface epithelium. In non-dysplastic tissue, only 24% of patients express cyclin A at the mucosal surface compared with 59% of patients with low-grade dysplasia, 87% of patients with high-grade dysplasia and 100% of patients with adenocarcinoma.35
Cyclin/Cdk inhibitors
The tumour suppressor p27 inhibits cyclin E/Cdk2 complexes, blocking cell cycle progression into S phase. p27 knockout mice have increased risk of oesophageal cancer compared to wild type mice and low levels or absence of p27 are associated with a worse prognosis in human colon, stomach, lung and prostate cancers.36 37 In Barrett's oesophagus and oesophageal adenocarcinoma lack of p27 expression is associated with malignant transformation and a poorer prognosis.38 In non-dysplastic Barrett's mucosa p27 expression is nuclear but in dysplastic mucosa, staining is often cytoplasmic. Low levels of p27 expression correlate with higher histological grade, depth of invasion and lymph node metastasis in patients with oesophageal adenocarcinoma.39 Nuclear localisation of p27 is essential for its growth-inhibiting function, and loss of expression or altered localisation in adenocarcinoma are associated with tumour progression and adverse prognosis, suggesting that p27 has a role in preventing progression of Barrett's epithelium to adenocarcinoma.
Insensitivity to anti-growth signals
Normal cell growth is restrained by inhibitory signals that block proliferation by inducing quiescence or permanent growth arrest (cellular senescence). Most anti-growth signals are controlled by the retinoblastoma gene protein (p185 Rb) at the G1 checkpoint. However, tumour cells can overcome this inhibition by inactivating tumour suppressor genes via mutation, allelic deletion (LOH) or promoter hypermethylation. Loss of the retinoblastoma gene itself seems to be rare in Barrett's metaplasia, but abnormalities in genes such as CDKN2A (encoding p16) and TP53, which normally block Rb phosphorylation and its activation, are relatively frequent.
p16
p16 (INK4 or CDKN2A) is a tumour suppressor gene on chromosome 9p21. p16 protein binds to and inhibits Cdk4/6, resulting in reduced phosphorylation of the retinoblastoma protein and inhibition of cell cycle progression through G1. Many studies have analysed p16 in cancers but fewer have examined premalignant lesions. Paulson and colleagues40 suggest that inflammation caused by exposure of oesophageal mucosa to acid and bile is a potential source of oxidative damage. Reactive oxygen and nitric oxide species may mediate mutations, including inactivation of p16, with subsequent uncontrolled cellular proliferation and disease progression.40 Early LOH appears to be a common mechanism of p16 inactivation41 associated with subsequent clonal expansion along the Barrett segment, favouring further mutations and facilitating disease progression.42 Other genetic and epigenetic events leading to loss of p16 include hypermethylation of CpG islands or allelic deletions. Immunohistochemistry has shown abnormalities of p16 expression in all grades of dysplasia. In Barrett's mucosa without dysplasia, p16 staining is nuclear. As dysplasia progresses, nuclear staining wanes while cytoplasmic positivity increases; an early signal and a potential mechanism of further genetic changes.43 A prospective study has shown that 9pLOH, 17pLOH and aneuploidy together predict progression to adenocarcinoma,44 but further studies are needed.
Avoidance of apoptosis
TP53
Neoplastic cells must avoid apoptosis to expand their numbers. Loss of p53 allows cells to bypass apoptosis and proliferate. TP53 is a tumour suppressor gene that encodes the protein (p53) involved in regulation of cell cycle progression, DNA repair, cellular senescence and apoptosis. It induces expression of p21 and mediates G1 and G2/M arrest. p53 and p21 prevent cells with DNA breaks from entering DNA synthesis, holding them back until they are repaired, or if repair is not possible, directing them to undergo apoptosis.45 p53 has a central role in human malignancy, being mutated in at least 50% of all malignant tumours.46 Mutations in TP53 have been reported in primary oesophageal adenocarcinomas and high-grade Barrett's mucosa, in which both alleles are lost, one by point mutation (90%) and the second by LOH.46 LOH refers to the loss of normal function of the other allele of a gene when the first allele is already inactivated. Point mutations of TP53 in oesophageal adenocarcinomas are often G:C to A:T transitions resulting from endogenous mechanisms such as exposure to oxygen and nitric oxide radicals.
Mutations in TP53 frequently increase the half life of p53, leading to increased levels of protein expression that can be detected by immunohistochemistry as unusually intense nuclear staining. In contrast normal (wild type) p53 has a short half life and is not readily detectable at all or is detected at low levels only.47 Although p53 mutations are common in adenocarcinoma, they are relatively uncommon in non-dysplastic Barrett's oesophagus and low-grade dysplasia. Patients with high-grade dysplasia often overexpress p53, suggesting that TP53 mutation may play a role in the transition from low-grade to high-grade dysplasia.48 49 Younes et al showed that p53 accumulation increased along the metaplasia- dysplasia-carcinoma sequence (0%, 9% and 87% for no dysplasia, low-grade dysplasia (LGD) and high-grade dysplasia (HGD)). On follow-up studies, only 1 of 21 patients with p53-negative biopsies developed dysplasia.
Overexpression of p53 may therefore be a marker of progression in patients histologically indefinite for dysplasia or with LGD only.50 Sikkema et al showed that p53 overexpression and Ki67 were predictive of progression from metaplasia to cancer.51 However, some TP53 mutations produce a truncated p53 protein undetectable by immunohistochemistry.47 Therefore, protein expression is neither as sensitive nor as specific as gene analysis. Coggi et al showed that in patients with p53 mutations, there was no detectable accumulation by immunohistochemistry in 31% cases.52 In addition, inflammation, DNA damage and other cellular stresses can upregulate p53. So, not all p53 mutations result in p53 protein accumulation, and not all protein accumulation is due to mutations.47 In an attempt to overcome some of these difficulties, a study of 325 patients with Barrett's oesophagus investigated the prevalence of 17pLOH as a marker of dysplasia and risk of progression to cancer.53 The prevalence of 17pLOH was 6% in non-dysplastic Barrett's mucosa, 57% in HGD, and it was an independent predictor of progression to adenocarcinoma. Of patients with baseline 17p LOH, 37% developed cancer whereas only 3% without 17p LOH progressed to cancer. These TP53 mutations seem to confer advantage to the mutant clone via three mechanisms: suppression of apoptosis, prevention of cell cycle arrest and senescence, and permitting genetic instability.54 Despite some of the limitations associated with the p53 protein expression, p53 is a well studied potential marker of neoplastic progression in Barrett's epithelium and newer genotyping technology may overcome some of the current limitations surrounding p53.
Nuclear factor κB (NFκB)
Transcription factor NFκB regulates proinflammatory genes, differentiation and growth. It exists in the cytoplasm of most cells in an inactive form complexed to the inhibitory molecule IκB that prevents the migration of the heterodimer to the nucleus. Cytokines, oxygen free radicals and acid stimulate translocation of NFκB to the nucleus, where it binds specific DNA sites and upregulates transcription of genes involved in inflammatory processes and immune responses.55 NFκB has been linked to lung fibrosis, autoimmune arthritis and irritable bowel disease (IBD).56
Over the years there has been much interest in the role of inflammation, either local or systemic, in the development of cancer. The NFκB pathway is therefore of interest in Barrett's epithelium, where there is often an associated inflammation. O'Riordan et al showed a stepwise increase in expression of NFκB, IL-8 and IL-1β in patients with Barrett's mucosa adjacent to adenocarcinoma.57 They also showed that NFκB was upregulated in 60% of patients with Barrett's oesophagus (all grades). In those with metaplasia but no dysplasia, 50% had NFκB overexpression; this rose to 63% in LGD and 100% in HGD. Patients with adenocarcinoma with increased expression of NFκB had elevated levels of cytokines IL-8 and IL-1. Further studies are required to determine the role of these molecules in the metaplasia-carcinoma sequence.
Cyclo-oxygenase 2 (COX-2)
COX-1 and the oncogene COX-2 cyclooxygenases mediate synthesis of prostaglandins from arachidonic acid. COX-1 is generally expressed whereas COX-2 is undetectable in most tissues. It is induced by cytokines, gastric acid and bile acids. Overexpression of COX-2 in vitro has effects from increasing cell proliferation, reducing apoptosis, promoting angiogenesis, decreasing E-cadherin expression and increasing the invasive and malignant potential of cells.42 58 COX-2 is detectable in metaplastic Barrett's mucosa and is overexpressed in high-grade dysplasia and adenocarcinoma. Its expression in LGD is similar to that of metaplasia in the absence of dysplasia.59 Other studies have reported a progressive increase in COX-2 expression along the metaplasia-dysplasia sequence.60 Different techniques have been used to evaluate COX-2 expression such as immunohistochemistry, western blotting and PCR, with inconsistent results. While COX-2 overexpression may play a role in Barrett's oesophagus, at present there is not enough data to support a useful role as a biomarker.
Invasion and metastasis
The Wnt signalling pathway
Wnt signalling is a key pathway in normal human organogenesis, but aberrant activation is implicated in carcinogenesis. Key genes and proteins in this pathway include the adenomatous polyposis coli (APC) gene, β-catenin and E-cadherin and although much is known about these molecules, mechanisms by which they interact are still incompletely understood. APC protein contains β-catenin degradation sites and E-cadherin has β-catenin binding sites. Disturbance of normal interactions between these molecules can lead to loss of growth inhibition or increased tumour invasiveness.61
β-Catenin
β-Catenin mediates cell–cell adhesion via the transmembrane glycoprotein E-cadherin. In carcinomas loss of the E-cadherin–catenin complex, which is involved in the maintenance of epithelial integrity, may confer increased invasiveness and metastatic ability on malignant cells.62 β-Catenin is also an oncoprotein that can lead to carcinogenesis when APC–β-catenin–T cell factor/lymphoid enhancer factor (TCF/LEF) signalling is disrupted. This is the so-called canonical Wnt signalling pathway.63 When a mitogenic Wnt signal from outside the cell is absent, glycogen synthase kinase 3 (GSK3) is active and, with APC and axin, binds to free β-catenin. In this complex, GSK3 phosphorylates β-catenin leading to its dissociation and breakdown in the cytoplasm.
The other mechanism inactivating β-catenin is its sequestration at the cell membrane by E-cadherin. In both cases, entry of β-catenin into the nucleus is prevented. In response to extracellular Wnt ligands acting on Frizzled receptors, or mitotic signalling, GSK3 activity is inhibited. As a result β-catenin is no longer broken down, resulting in an excess of free β-catenin. Its cytoplasmic concentration rises, binding to other transcription factors (such as TCF/LEF) and it is transferred to the nucleus. Nuclear proteins in combination with β-catenin lead to activation/suppression of individual gene promoters, and to cellular proliferation.61
Nuclear β-catenin staining is absent in Barrett's metaplasia without dysplasia but is frequently strong in adenocarcinomas. Membranous expression of inactive β-catenin changes with progression from metaplasia to adenocarcinoma with reduced expression seen in 80% of adenocarcinomas, 68% HGD, 16% LGD and 5% IM without dysplasia.64 There are no obvious mutations in β-catenin itself, implying perhaps that other alterations in Wnt signalling may be important.65
E-Cadherin
E-Cadherin is a transmembrane protein essential for maintenance of cells during development. The extracellular domain of E-cadherin mediates adhesion with cadherins on neighbouring cells, while the intracellular domain interacts with cytoplasmic proteins linked to actin via catenins. It plays a crucial role in cell–cell adhesion and reduced expression is an important molecular event concerned with invasion and metastases.66 Along with β-catenin several studies have shown decreased E-cadherin expression with progression from Barrett's metaplasia to adenocarcinoma.41 63 Reduced E-cadherin expression promotes epithelial cell invasiveness and metastasis in various human cancers.67 Recent studies have shown that E-cadherin has aberrant nuclear localisation in some tumours (pancreatic endocrine tumours, oesophageal squamous and colorectal cancers.68 69 Further research is required into the role of Wnt signalling in oesophageal adenocarcinoma.
Ploidy
Aneuploidy (abnormal cellular DNA content) is associated with increased risk of progression to dysplasia and adenocarcinoma. A large continuing phase 4 study by Reid et al has shown that patients with no dysplasia, indefinite or low-grade dysplasia at baseline biopsy, and a diploid cell population (no aneuploidy) are at low risk of progression to adenocarcinoma.70 Patients in whom baseline biopsies demonstrated aneuploidy, tetraploidy (4N DNA content) or high-grade dysplasia had 5 year cancer incidences of 43%, 56% and 59%, motivating more intense surveillance. In some centres, such as Seattle, flow cytometry to assess aneuploidy is routinely undertaken in the assessment of Barrett's biopsy samples.
Despite good evidence in favour of ploidy as an early risk marker in Barrett's oesophagus, it is little used in clinical practice, probably because of its requirements in terms of costs and instrumentation and reimbursement issues. Flow cytometry has the disadvantage of divorcing DNA content measurements from morphology. Image cytometry of intact nuclei from thick sections partly addresses this issue by allowing histological control of the material submitted for analysis. Image cytometry on histological sections gives the best correlation of morphology and DNA content measurements but introduces its own problems with nuclear truncation and overlapping. Nevertheless, DNA content measurements are possible on sections around 7 microns thick, and further evaluation in this area would be desirable. Fleskens et al show that combining DNA content measurement with the cell cycle marker Ki67 facilitates detection of aneuploid cell populations in oral premalignancy, and demonstrate combined immunofluoresence and staining with a fluorescent DNA intercalating agent (DRAQ5) under stoichiometric staining conditions.71
α-Methylacyl-coenzme A racemase (AMACR)
AMACR is a protein expressed in peroxisomes and mitochondria of normal liver and kidney cells, and plays a role in the β-oxidation of branched chain fatty acids.72 AMACR overexpression was initially reported in prostate cancer and high-grade intraepithelial neoplasia but it is also expressed in dysplastic cases of Barrett's oesophagus.73 Immunohistochemistry suggests AMACR is not expressed in non-dysplastic Barrett's epithelium, but is present in low-grade dysplasia (38%), high-grade (81%) and adenocarcinoma (72%).72 The exact role of AMACR in the oesophageal epithelium is unclear but its overexpression may be a useful adjunct in diagnosing dysplasia in difficult cases. Future studies are required to fully explore the role of AMACR and its prognostic significance.
Conclusions
Barrett's ‘columnar lined’ oesophagus is important as the precursor of oesophageal adenocarcinoma, which has the most rapidly rising incidence of any solid tumour in the Western world, with Scotland having rates as high as anywhere.1 Advances in disease management over the last decade have seen improvements in endoscopic therapies to treat high-grade dysplasia, better imaging and biopsy detection systems, and several candidate molecular biomarkers. At present, dysplasia develops in around 5% of patients with Barrett's oesophagus, with 10% to 50% progressing to high-grade dysplasia and cancer over 2–10 years. The remainder remain static. Despite the risk of malignant progression, only 2% to 3% of patients with Barrett's oesophagus will die from oesophageal adenocarcinoma, and overall life expectancy is not very different from those without the disease.
The role of biomarkers in Barrett's oesophagus is potentially twofold. First, to identify patients at risk of progression to high-grade dysplasia and cancer, so they can be diagnosed and treated earlier with endoscopic therapies, minimising morbidity and avoiding the morbidity and mortality of oesophagectomy. Second, and almost equally useful, markers able to identify patients at little or no risk of progression would allow less frequent surveillance endoscopy and biopsy for low-risk patients, minimising healthcare costs and patient anxiety. Being able to reassure a patient of a low progression risk is at least as important as to be able to assign a high risk, given limited evidence of effective risk management.
Barrett's oesophagus is a complex disease process with significant genetic heterogeneity, and greater heterogeneity identified within a Barrett's segment is itself a predictor of disease progression.74 Many individual mutations have been identified, but no single marker has yet been identified with ideal characteristics or the potential to fulfil clinical requirements on its own. It may be naïve to expect a single biomarker will fulfil all expectations in such a complex disease and many centres now think biomarker panels may be more likely to aid management.
Dysplasia, our ‘gold’ standard biomarker, and aneuploidy are at present the only markers routinely used in clinical practice. Many biomarkers have not passed through phase 3 or 4 trials and much more work needs to be performed in this area before any of them is established on secure evidence as a basis for clinical practice. Multicentre trials will be required for assessment and integration of clinical and molecular variables so comprehensive conclusions can be made. This will require a degree of cooperation rarely so far seen in the field, but without which greater understanding and appropriate management of Barrett's oesophagus will be further delayed.
Take-home messages
Barrett's oesophagus is a premalignant condition for the development of oesophageal adenocarcinoma. The diagnosis and management of this condition currently depend on the presence and degree of dysplasia. Dysplasia, especially low grade, can be difficult to identify and the search for a clinically useful and predictive biomarker continues. Aneuploidy is a predictive marker for disease progression but further work is required before used in routine clinical practice.
Interactive multiple choice questions
This JCP article has an accompanying set of multiple choice questions (MCQs). To access the questions, click on BMJ Learning: take this module on BMJ Learning from the content box at the top right and bottom left of the online article. For more information please go to: http://jcp.bmj.com/education. Please note: the MCQs are hosted on BMJ Learning the best available learning website for medical professionals from the BMJ Group. If prompted, subscribers must sign into JCP with their journal's username and password. All users must also complete a onetime registration on BMJ Learning and subsequently log in (with a BMJ Learning username and password) on every visit.
References
Footnotes
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.