Article Text

Abstract
Birt–Hogg–Dubé syndrome (BHD) is an autosomal dominant inherited disorder characterised by fibrofolliculomas, renal tumours, pulmonary cysts and pneumothorax. The pulmonary cysts and repeated episodes of pneumothorax are the clinical hallmarks for discovering families affected by the syndrome. This disorder is caused by mutations in the gene coding for folliculin (FLCN). FLCN forms a complex with FLCN-interacting protein 1 (FNIP1) and FNIP2 (also known as FNIPL), and the complex cross-talks with signalling molecules such as 5′-AMP-activated protein kinase (AMPK) and the mammalian target of rapamycin (mTOR). Heterozygous Flcn knockout mice and rats with Flcn gene mutations develop renal cysts, adenomas and/or carcinomas. These findings suggest that FLCN functions as a tumour suppressor that inhibits renal carcinogenesis. However, the mechanisms of the formation of pulmonary cysts and pneumothorax associated with heterozygous mutations in FLCN are poorly understood. Resected lung specimens from patients with BHD are often misdiagnosed by pathologists as non-specific blebs or bullae or emphysema, and patients with BHD who have pulmonary cysts and repeated pneumothorax frequently do not receive appropriate medical investigations. This review discusses the clinical and pathological features of lungs of patients with BHD, focusing on the diagnostic pathology and possible mechanisms of cyst formation.
- Familial Cancers
- Molecular Pathology
- Pulmonary Pathology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Birt–Hogg–Dubé syndrome (BHD) is an inherited disorder characterised by multiple fibrofolliculomas, pulmonary cysts, pneumothorax and renal cysts and tumours. In 1977, Birt, Hogg and Dubé investigated members of a Canadian family who presented with thyroid cancers and found that some members of the kindred had fibrofollicular skin tumours that occurred in an inherited autosomal dominant pattern.1 Although the familial disorder was later named the Birt–Hogg–Dubé syndrome, similar inherited skin tumours had been reported 2 years previously. The German dermatologists Hornstein and Knickenberg described a middle-aged woman and her brother who had multiple perifollicular fibromas and intestinal polyps.2 They reported the detailed clinicopathological features of the siblings and suggested that the disorder was probably inherited from their father who had bilateral renal cysts and unilateral lung cysts in addition to skin tumours. Hornstein and Knickenberg called the systemic disorder ‘a cutaneo-intestinal syndrome sui generis’.2 The authors of both reports discussed the possibility of a distinctive hereditary disorder and referred to several reports on similar skin diseases that had been published before 1975.1 ,2 Birt et al commented that the histopathological features of the skin tumours described by Hornstein and Knickenberg were different from those that they had characterised. However, dermatopathologists have subsequently concluded that the fibrofolliculomas, perifollicular fibromas, trichodiscomas and acrochordons seen in patients with BHD represent a spectrum of the same skin tumour. In a recent review, Happle insisted that the works by Hornstein and Knickenberg should be recognised and proposed that the syndrome should be renamed ‘Hornstein–BHD’.3 We would like to comment briefly on this issue. If the syndrome is renamed, we believe that Knickenberg should also be included. There is no justification for omitting the name of the woman physician who also contributed greatly to the series of studies in the 1970s. Both Birt et al and Hornstein and Knickenberg referred to previous studies reporting skin tumours associated with familial occurrence or systemic disorder. It is possible that other authors concerned with crediting the investigators responsible for first reporting on patients with clinical manifestations of this syndrome may have different priorities with regard to proper attribution.
In 2001, two groups of investigators found the chromosomal location of the gene responsible for BHD.4 ,5 One year later, Nickerson et al delineated the susceptibility locus of the gene on chromosome 17p11.2.6 The gene was called BHD at first. At present, the official symbol referring to the gene is called FLCN (folliculin) (OMIM: 607273). FLCN consists of 14 exons (NCBI Reference Sequence: NM_144997.5).
Among the characteristic manifestations of patients with BHD, renal cell carcinoma (RCC) is the most serious because of its poor prognosis. RCC generally occurs in adult members of a BHD-affected family and rarely affects infants and teenagers. Pulmonary cysts and pneumothorax are found occasionally in young adult patients aged 20–30 years. Skin tumours also develop around the age of 30 years, whereas RCC tends to affect middle-aged and elderly individuals with the syndrome.7 It is therefore important to correctly diagnose patients who visit hospitals with repeated pneumothorax and those who have fibrofolliculomas in visible regions such as the neck and face (figure 1C,D). The pulmonary manifestations, however, are frequently misdiagnosed as spontaneous blebs, bullae or some other emphysematous condition.8–10 The pathological features of the lung in patients with BHD have not been described in the major textbooks on pulmonary disease so pathologists still have problems distinguishing BHD-associated pulmonary cysts from blebs and bullae. The mechanisms of cyst development are largely unknown. In this review we describe in detail the pathological features of the pulmonary cysts and ruptured lesions found in lung tissue of patients with BHD. Diagnostic clues and recent advances in the molecular studies of FLCN-mediated signalling are also presented.

Thoracic CT scans and skin tumours of patients with Birt-Hogg-Dubé syndrome. (A) A 33-year-old woman who had a pneumothorax six times showing cysts localised in the perimediastinal subpleura. (B) A 53-year-old woman who had a pneumothorax twice showing most of the cysts in contact with the interlobular septum. (C) A 69-year-old woman with renal cell carcinoma who had multiple skin papules on the neck. (D) A 76-year-old man with a few flat-topped fibrous papules on the back (arrows).
Clinical features of pulmonary cysts in BHD
Epidemiological studies have demonstrated that multiple pulmonary cysts and multiple episodes of pneumothorax are frequently observed in patients with BHD.11 Imaging studies using thoracic CT have found high rates of multiple pulmonary cysts in more than 80% of BHD-affected families.11 ,12 In our practice, all 20 patients genetically diagnosed with BHD were found to have multiple pulmonary cysts on thoracic CT (figure 1A,B). Several differential diagnoses should be considered in patients who have a history of repeated pneumothorax, especially in women of reproductive age. Lymphangioleiomyomatosis (LAM) and pulmonary endometriosis should be included in the differential diagnosis. LAM tends to occur in women of reproductive age and about 30% of women with LAM have tuberous sclerosis complex (TSC),13 ,14 which will be discussed later. Other genetic disorders such as α-antitrypsin deficiency and Marfan syndrome can also be considered.
Investigating the possible association between pulmonary BHD and chronic obstructive pulmonary disease (COPD), Cho et al compared single nucleotide polymorphisms (SNPs) of FLCN. None of those SNPs in patients with COPD were the same as the mutations previously reported for BHD, and there was no statistical association for four selected variants with the presence of COPD or emphysema-related phenotypes. Based on their results, they concluded that BHD and COPD may have distinct genetic causes.15
Thoracic CT yields findings that greatly aid in the diagnosis of BHD. Tobino et al summarised the detailed radiological findings of the pulmonary lesions in 12 patients with BHD.16 ,17 Many pulmonary cysts occurred in individual patients; in a single patient with severe disease there were 407 pulmonary cysts bilaterally located in the medial and subpleural regions. The lower part of the middle zone was more frequently affected than the apex of the lung. Similar findings were reported by Ayo et al who reviewed five cases.18 In our practice, we also observed similar radiological features in 19 patients with pulmonary cysts and/or pneumothorax who were diagnosed by genetic testing. Cysts occurring in the middle and lower lobes towards the mediastinum and intimate association of the cysts with interlobular septa and/or visceral pleura are therefore characteristic CT findings that aid in differentiating BHD lesions from LAM and other cystic pulmonary lesions.17
Pathological features of pulmonary cysts
Very little information on the microscopic findings of pulmonary cysts in BHD is available.19–21 Some case reports of patients with BHD have described pathological findings that were not reliable because the lung specimens were inadequate.8–10 In these reports, BHD-associated pulmonary cysts were frequently confused with blebs, bullae or other cystic air space diseases. The pathological findings previously reported were based on ruptured lesions that caused pneumothorax. Only fragmented pleural walls were resected in these cases and detailed microscopic observation was not available. These specimens had elongated visceral pleurae with hyalinised stroma and, in some specimens, the tissue included true blebs or bullae with mesothelial invagination. It was inevitable that these specimens containing BHD-associated lung lesions were found to have non-specific blebs or bullae; however, physicians and pathologists should have been sceptical about the diagnosis because there were multiple bilateral cystic lesions, repeated clinical episodes and positive family histories. These cases should be carefully re-examined.
In unruptured pulmonary cysts associated with BHD, the cyst wall expands towards the visceral pleura and is partially incorporated into the parenchyma, interlobular septum and/or bronchovascular bundle (figure 2A). Enlarged cysts can also appear segmented by an alveolar wall and resemble a multicystic pneumatocoele and become deeply embedded in the interlobular septum (figure 2B).19 The inner surface of the cyst is lined by alveolar cells immunostaining for epithelial markers and surfactant proteins (figure 2C). These epithelial cells do not show neoplastic proliferation or atypical morphology. They may be attenuated or not easily visible; however, cuboidal cells resembling type II pneumocytes are often observed in the innermost layer. Sometimes a cyst wall is partially enveloped by another wall, demonstrating the unique pattern of ‘an alveolus or a few alveoli within a cyst’ (figure 2D). Some cysts have veins protruding into the cystic space (figure 2E). These characteristic findings are important microscopic clues for the pathological diagnosis.20 Most of the epithelium lining the cyst appears to be friable. Some epithelial tissue exfoliates from the cyst wall and some remaining tissue appears thin; detailed evaluation shows unusual alveolar architecture (figure 2D) and epithelial cells budding in the walls of the cyst in a few cases (figure 2F). These complex structures are often associated with chronic inflammatory cell infiltration of various degrees, which suggests a possible development of cysts due to inflammation. We speculate that these might resemble hamartomatous cysts that expand very slowly, modified in their structures by inflammation, and sometimes rupture, causing pneumothorax.
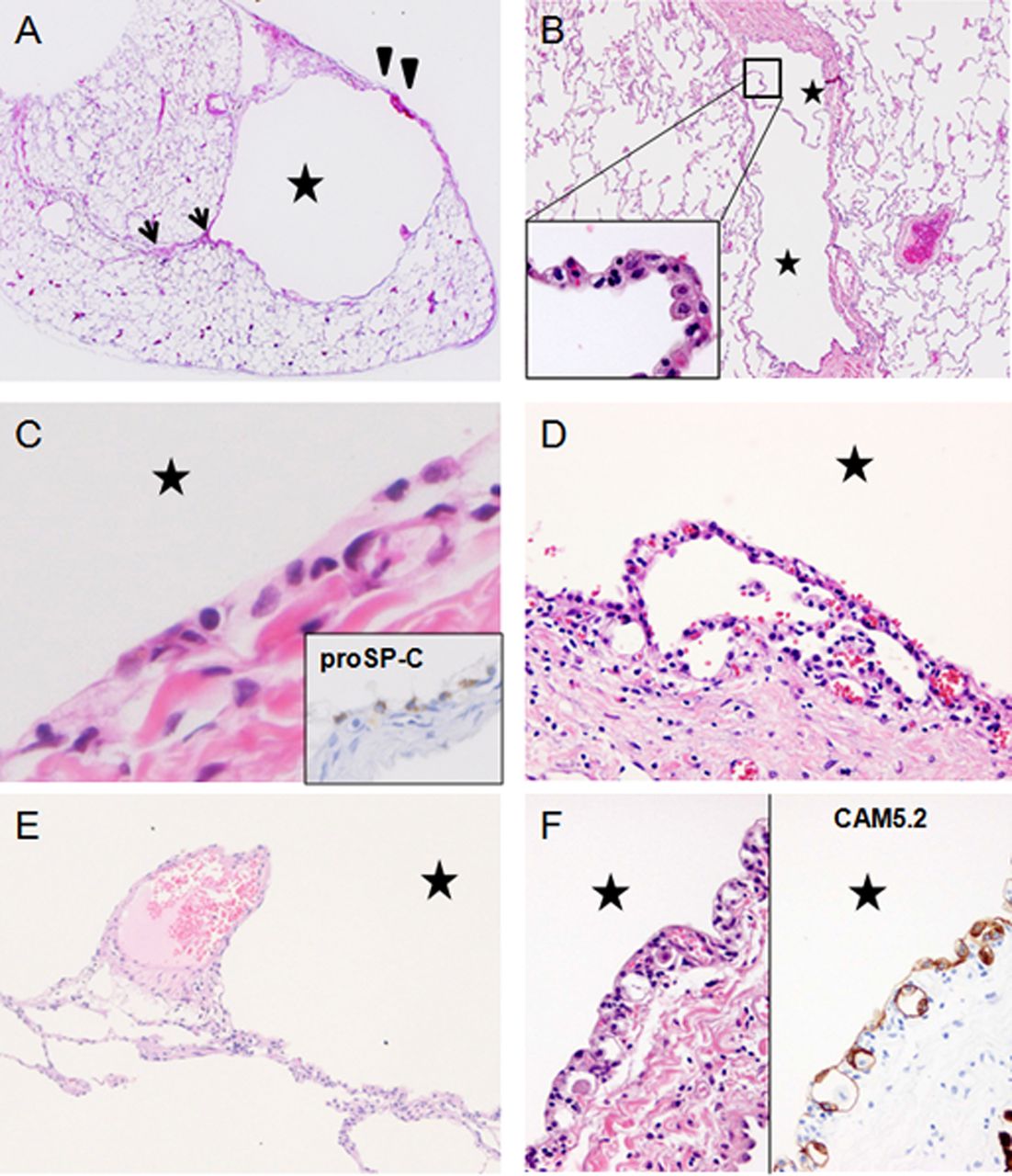
Histopathological features of lungs affected by Birt–Hogg–Dubé syndrome (BHD): H&E staining of resected lungs from patients diagnosed with BHD by genetic testing. Cysts are indicated by stars. (A) A resected cyst from a 41-year-old woman. The cyst wall partially incorporates the pleura (arrowheads) and interlobular septum (arrows). (B) A double-spaced microscopic cyst from the same patient. The inner surface is lined by pneumocytes (inset) and is partially embedded in an interlobular septum (reproduced from Koga et al19 with permission of the publisher). (C) Innermost layer of a cyst: the lining cells show neither aberrant proliferation nor pleomorphic features. They are immunostained for proSP-C, suggesting pneumocytes. (D) A few alveoli abut on the innermost layer and are anastomosed to the cyst lining cells. (E) Finger-like protrusion of a vein into the cystic space. (F) Epithelial spores lining the inner layer of the cyst wall (left), which is highlighted by immunostaining for cytokeratin CAM5.2 (right).
In pneumothorax-associated ruptured cysts and cysts before rupture, the histopathology becomes complicated because of mechanical stress and tissue remodelling with associated inflammation. Pleural thickening, interstitial bleeding and hyalinisation alter the original architecture of the cysts. After rupture, mesothelial invagination and bleb formation are frequently observed (figure 3A,B). Cuboidal cells resembling type II pneumocytes tend to be more conspicuous at this stage. These metamorphosed pleurae demonstrate the non-specific histopathology common to blebs and interfere with the correct diagnosis. We therefore suggest that surgeons who perform pulmonary wedge resections in patients suspected of having BHD should also sample cystic areas that are not ruptured. Cystic alveoli and fusion of the epithelium of the cyst to the mesenchyme are diagnostic clues to BHD-associated lung lesions even at advanced stages with inflammatory modifications. The interlobular septum in contact with the cyst is sometimes oedematous (figure 3C, arrows), which may be a result of localised disturbance of the circulation and venous stasis. Other possible causes of oedema include FLCN insufficiency, which might lead to matrix remodeling,22 or angiogenic factors that increase vascular permeability (see later).

Histopathological features of ruptured cysts and hypothetical mechanism of cyst growth. (A) Ruptured visceral pleura thickened with fibrosis and inflammation. Mesothelial invagination (arrowheads) is observed. Inset shows the invaginated cells immunostained for calretinin. (B) Elongated cysts form a complicated structure. (C) Interlobular septum (arrows) associated with a cyst shows oedematous features. (D) The cyst lining epithelial cells are strongly immunostained for p-mammalian target of rapamycin (mTOR) (left) and p-S6 (right). (E) A hypothetical signalling pathway involved in pulmonary cyst growth. Folliculin insufficiency in cyst composing cells leads to mTOR activation and accelerates downstream molecules such as p-S6 and vascular endothelial growth factor (VEGF). FNIP, FLCN-interacting protein; HIF-1α, hypoxia-inducible factor 1α; mTORC1 and 2, mTOR complex 1 and 2.
Table 1 summarises the histopathological features that help to differentiate between BHD-associated cysts and idiopathic blebs and/or bullae. Small unruptured BHD cysts generally are free of signs of inflammation and fibrosis. Histopathological observations have indicated that BHD-associated cysts are initially located very close to the interlobular septa and/or are subpleural. Some cysts, if not all, become larger and prone to rupture, leading to pneumothorax. Ruptured cysts or modified cysts before rupture are accompanied by inflammation and become indistinguishable from blebs or bullae because of pleural fibrosis and hyalinisation. These metamorphosed cyst walls, however, may still retain some of the unique features mentioned above (figure 2D,F).20 Modified BHD-associated pulmonary cysts may secondarily form blebs and/or bullae, but not vice versa.
Comparison of the lung cystic lesions between Birt–Hogg–Dubé syndrome (BHD) and blebs/bullae
Several disorders other than blebs/bullae should be considered in the differential diagnosis of BHD-associated cysts including emphysema, infectious lung diseases, LAM and thoracic endometriosis. Immunohistochemical staining using the monoclonal antibody HMB45 for melanoma-related antigen is helpful for differentiating between LAM- and BHD-associated cysts; however, in the early stages of LAM, HMB45-positive cells are inconspicuous. Female patients with BHD who have repeated episodes of pneumothorax are more likely to be carefully investigated than young male patients with BHD who tend to be misdiagnosed with idiopathic pneumothorax. Comprehensive information that includes clinical and family histories can alert the physician to the possibility of BHD. On the other hand, long-term smoking or long-term smoking-associated emphysematous disorder may mask the pulmonary microscopic characteristics of BHD, especially in elderly patients.
It should be noted that there is cystic lung disease of unexplained cause.23 We also have had some patients who could not be diagnosed. These patients had family members with pneumothorax and/or histories of repeated episodes of pneumothorax. The radiological and histological features of the lungs of these patients were indistinguishable from the features of BHD-affected lungs; however, neither FLCN mutations nor intragenic deletions and duplications were identified in these patients. Investigations are needed to identify more specific diagnostic clues for diagnosing BHD in affected lungs and to understand the pathophysiology of non-BHD-associated pulmonary cysts that share some features with BHD-associated cysts.
Symptoms associated with dysfunctional FLCN
FLCN is regarded as a tumour suppressor and its dysfunction leads to renal carcinogenesis. However, the role of FLCN in human pulmonary cysts is poorly understood. Flcn+/− rodent models for BHD are not good for demonstrating pulmonary cysts.24 ,25 A lung-targeted Flcn-depleted animal model has not yet been developed. Sequence analysis of FLCN from microdissected fibrofolliculomas demonstrated a heterozygous pattern,26 but FLCN in the pulmonary cysts of patients with BHD has not been characterised because the cells lining the cysts do not show neoplastic proliferation and the numbers of epithelial cells are too small for analysis. A histological study using in situ hybridisation reported that FLCN mRNA was detectable in stromal cells and type I pneumocytes in the lungs of patients with BHD.27 In our immunohistochemical study, macrophages, pneumocytes and even the cells lining the cysts of BHD-affected lungs stained positive for FLCN,19 ,20 whereas there were many unstained RCC cells in BHD-affected kidneys.20 These data suggest that FLCN in pulmonary cysts probably displays haploinsufficiency.
Little is known about the metachronicity of pulmonary cyst development. Many cysts appear to remain unruptured because the frequency of pneumothorax in elderly patients with BHD is not as high as the frequency of pneumothorax in middle-aged patients. Thus, some cysts may evolve continually and rupture at different points in time. Alternatively, abnormal epithelial/mesenchymal interactions may weaken the extracellular matrix of the visceral pleura leading to a pneumothorax-prone condition. BHD-associated skin tumours demonstrate proliferation of mesenchymal cells in addition to extension of the follicular epithelium, indicating that dysfunctional FLCN affects both cell types. FLCN mRNA is detectable in both the epidermal and stromal components of the skin.27 Additional studies are needed for better understanding of the budding and development of pulmonary cysts and the process of pneumothorax formation in patients with BHD.
Studies of BHD rodent models have elucidated the importance of FLCN in renal carcinogenesis. Flcn-null mice and rats are embryonic lethal, and Flcn heterozygous knockout mice and rats with Flcn mutations develop renal cell adenomas and RCC.28 ,29 Nihon rats with heterozygous Flcn mutations develop renal cell adenomas at around 3–4 weeks which transform to RCC when the rats are 6 months old.30 The carcinoma cells of RCC in this model frequently show loss of heterozygosity of Flcn. Somatic second hit mutations of Flcn have been found in RCC of patients with BHD.31 The BHDf/d/KSP-Cre mouse has kidney epithelium-specific depletion of Flcn, develops polycystic tubules within 2 weeks after birth and dies at 3 weeks from renal failure.32 These results indicate that FLCN works as a tumour suppressor and also as a possible regulator of cystic remodelling.
In heterozygous Flcn knockout mice (Flcn+/− mice), adenocarcinoma and adenoma of the lung are detected sporadically.24 A few human cases of low-grade atypical adenomatous hyperplasia and lung carcinomas have been reported,7 ,18 and we also had a patient who developed bronchioloalveolar carcinoma with multiple pulmonary cysts (figure 4). There is a recent report of a 50-year-old man with BHD who was found to have a 12 mm histiocytoma in the lung plus multiple cysts and pneumothorax.33 It is unknown whether the lung carcinoma and other types of lung tumours in patients with BHD are associated with FLCN haploinsufficiency. Long-term prospective studies are needed to elucidate whether neoplastic changes in the lung can occur.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schemes of mutation patterns of 19 Asian families with Birt–Hogg–Dubé syndrome (BHD). The responsible mutation sites of the 19 Asian families are indicated. F6 is excluded because genetic testing was done in another institute. F5 is Taiwanese and the others are Japanese families with BHD. The most frequent mutations are 4 bp deletion in exon 13 (c.1533_1536delGATG) and duplication of cytosine in exon 11 (c.1285dupC).
FLCN-associated signalling pathways
FLCN is involved in the mammalian target of rapamycin (mTOR) signalling pathway.34 Recent studies revealed that FLCN forms a complex with two associated proteins. Baba et al identified the first binding protein of FLCN in 2006 and called it FLCN-interacting protein 1 (FNIP1). In 2008, they and other investigators identified a novel FNIP1 homologue, FNIP2 (also known as FNIPL).35 ,36 The FLCN complex interacts with 5′-AMP-activated protein kinase (AMPK)34–36 and regulates mTOR signalling. High levels of dysregulated mTOR activity are associated with several systemic syndromes including TSC. The complex of hamartin (TSC1) and tuberin (TSC2) interact with AMPK and mTOR, and mutations in TSC1 or TSC2 lead to uncontrolled cell growth.37 TSC is associated with characteristic neoplasms such as neurofibromas in the skin, angiomyolipoma in the kidney and LAM in the lung. Other mTOR-associated hamartoma syndromes include Peutz–Jegers syndrome and Cowden's disease in which respective heterozygous mutations in STK11/LKB1 and PTEN lead to hamartomatous colorectal polyps. BHD syndrome, TSC and the other mTOR-associated disorders have in common conditions such as multiple skin tumours and hereditary tumours and hamartomas which are associated with haploinsufficiency of the corresponding gene products. It is not completely understood whether the FLCN complex plays a role in mTOR signalling similar to the role of TSC1 and TSC2.
Both inhibitory and stimulatory effects of FLCN on mTOR complex 1 (mTORC1) have been seen, depending on the experimental design of the study,24 ,28 ,32 ,35 ,36 which indicates that the function of FLCN in mTOR signalling may be dependent on context.25 We have shown that the cells lining pulmonary cysts stain positively for phospho-mTOR and phospho-S6 expression (figure 3D).20 We also found that hypoxia-inducible factor 1α and vascular endothelial growth factor (VEGF) were immunostained strongly in lung specimens from patients with BHD (unpublished observation). Although statistical validation of a sufficient number of patients is not yet available, the results of our histopathological investigations suggest that mTOR signalling is accelerated in the cells lining pulmonary cysts, and that downstream molecules such as S6 protein and VEGF contribute to the development of cysts under conditions of FLCN haploinsufficiency (figure 3E). Recent studies have elucidated the suppressor activity of FLCN in the regulation of transcription factor E3 and transforming growth factor β which are widely involved in tumorigenesis and apoptosis.38 ,39 Further investigations of the molecular mechanisms involved in the formation of pulmonary cysts and rupture are needed.
FLCN mutations
In 2009, more than 50 germline mutations were reported from families affected by BHD40 and, at present, over 100 have been reported. The C8 tract in exon 11 is the most frequently affected site; duplication of cytosine (C9) and deletion (C7) have both been reported. A statistical association between mutations and clinical manifestations has not been clarified. Differential phenotypes are often observed in family members with BHD, and not all the members will develop RCC.19 Painter et al reported that all 24 affected members of a Finnish kindred had multiple pulmonary cysts on thoracic CT.41 They suggested that a 4 bp deletion in exon 4 (c.235_238delTCGG) causes pulmonary cysts with 100% penetrance. Toro et al investigated 189 patients with BHD with pulmonary cysts and pneumothorax and found that patients with FLCN mutations in exons 9 and 12 had pneumothorax more frequently than patients with FLCN mutations in other exons.12 Schmidt et al suggested that patients with cytosine deletion in the C8 tract of exon 11 (c.1285delC) may have a lower risk of developing RCC than individuals with cytosine duplication in the C8 tract (c.1285dupC).42 RCC tends to occur in elderly patients with BHD, thus long-term follow-up survey in age-matched groups is needed.
Although not all the kinships we studied underwent thoracic CT, our data indicate that a 4 bp deletion in exon 13 (c.1533_1536delGATG) causes pulmonary cysts with 100% penetrance (n=11 affected members from five different families; figure 4). Data are needed from more kinships to clarify statistical associations between the mutation patterns and clinical manifestations. According to the FLCN mutation online database (http://www.skingenedatabase.com/), the registered mutations are mainly from families in the USA. Kunogi et al summarised the mutations in Japanese pedigrees and compared them with those reported by the National Cancer Institute.43 There were differences in the frequencies of mutation sites. For example, a 7 bp duplication in exon 12 (c.1347_1353dupCCACCCT)19 ,43 and mutations of a splice acceptor site in intron 57 ,43 have not been reported from American and European pedigrees.7 In addition, we found a few other unique mutations in Asian pedigrees.20 ,44 Figure 4 depicts a summary of the mutations from 19 Asian families affected by BHD (18 Japanese and 1 Taiwanese) which we analysed between 2008 and 2012. A cytosine duplication or deletion in the C8 tract of exon 11 (c.1285dupC or c.1285delC) is known to be the ‘hot spot’ in Western patients with BHD. Mutation analyses by us20 and another Japanese group43 have added new information; in Japanese patients with BHD, c.1347_1353dupCCACCCT in exon 12 and c.1533_1536delGATG in exon 13 are the ‘hot spots’.
Conclusions and future prospects
Around 10 years have passed since the discovery of FLCN.6 Increasing numbers of case reports and reviews on BHD pedigrees have provided physicians with the diagnostic features of this inherited disorder.45 ,46 However, there are still many patients who are misdiagnosed and thought to have spontaneous pneumothorax and sporadic RCC. Pneumothorax and chest pain are clinically important signs and symptoms and warrant a thorough medical examination,47 and it is important to keep in mind that the onset of repeated pneumothorax generally starts at an earlier age than RCC in patients with BHD.11 Pathologists should carefully distinguish pulmonary lesions associated with BHD from the lesions that occur in other emphysematous diseases. In this review we have presented the diagnostic histopathological features including the diagnostic puzzles of pulmonary cysts. Pathologists must remember that resected lesions are frequently modified by mechanical stress and post-rupture remodelling. In addition, it is not completely understood how pulmonary cysts develop under conditions of FLCN insufficiency. A better understanding of the molecular and cellular cross-talks and meta-analyses of patients with BHD are needed to develop a more precise diagnosis and better follow-up of affected families.
Key messages
-
In un-ruptured pulmonary cysts associated with Birt-Hogg-Dubyndrome (BHD), the cyst wall expands toward the visceral pleura and is partially incorporated into the parenchyma, interlobular septum and/or bronchovascular bundle.
-
The inner surface of the cyst is lined by alveolar cells that may be attenuated or not easily visible; however, cuboidal cells resembling type II pneumocytes are often observed.
-
In pneumothorax-associated cysts, the histopathology becomes complicated because of mechanical stress and tissue remodeling with inflammation.
Acknowledgments
The authors thank members of the molecular pathology laboratory at the Yokohama City University Graduate School of Medicine for assistance, Dr R Tanaka and Dr T Matsuzawa for sequence analysis and other BHD-Net members for clinical data.
References
Footnotes
-
Funding This work is supported by Grants-in-Aid for Scientific Research (MF and YN) (Grant No. 24590408).
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/