Article Text

Abstract
Aims Mutation detection accuracy has been described extensively; however, it is surprising that pre-PCR processing of formalin-fixed paraffin-embedded (FFPE) samples has not been systematically assessed in clinical context. We designed a RING trial to (i) investigate pre-PCR variability, (ii) correlate pre-PCR variation with EGFR/BRAF mutation testing accuracy and (iii) investigate causes for observed variation.
Methods 13 molecular pathology laboratories were recruited. 104 blinded FFPE curls including engineered FFPE curls, cell-negative FFPE curls and control FFPE tissue samples were distributed to participants for pre-PCR processing and mutation detection. Follow-up analysis was performed to assess sample purity, DNA integrity and DNA quantitation.
Results Rate of mutation detection failure was 11.9%. Of these failures, 80% were attributed to pre-PCR error. Significant differences in DNA yields across all samples were seen using analysis of variance (p<0.0001), and yield variation from engineered samples was not significant (p=0.3782). Two laboratories failed DNA extraction from samples that may be attributed to operator error. DNA extraction protocols themselves were not found to contribute significant variation. 10/13 labs reported yields averaging 235.8 ng (95% CI 90.7 to 380.9) from cell-negative samples, which was attributed to issues with spectrophotometry. DNA measurements using Qubit Fluorometry demonstrated a median fivefold overestimation of DNA quantity by Nanodrop Spectrophotometry. DNA integrity and PCR inhibition were factors not found to contribute significant variation.
Conclusions In this study, we provide evidence demonstrating that variation in pre-PCR steps is prevalent and may detrimentally affect the patient's ability to receive critical therapy. We provide recommendations for preanalytical workflow optimisation that may reduce errors in down-stream sequencing and for next-generation sequencing library generation.
- PCR
- MOLECULAR PATHOLOGY
- diagnostic screening
- MELANOMA
- LUNG CANCER
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Use of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor erlotinib as first-line treatment for non-small cell lung cancer (NSCLC) illustrates the potential for targeted drugs as alternatives to chemotherapy.1–5 Initially, retrospective sequencing of tumours from responders revealed activating EGFR mutations, making tumour profiling a priority in NSCLC management.6 Similarly, vemurafenib and BRAF p.V600E have shaped workup of other cancers.7–9 This led to incremental improvements in PCR technology, culminating recently in PCR enrichment for next-generation sequencing (NGS) library making.10 Despite technical advances, assessing the accuracy of pre-PCR steps, which include DNA extraction from formalin-fixed paraffin-embedded (FFPE) tissues, DNA quantitation and DNA quality control, remains a key challenge in external quality assurance (EQA). Only two EQA programmes have previously assessed DNA extraction and quantitation in conjunction with downstream mutation detection; however, both of these tested DNA extraction from blood samples rather than FFPE.11
FFPE tissue preparation emerged 100 years ago as a method for long-term tissue preservation.12 Today, mutation analysis from FFPE-derived DNA assists diagnosis of most solid tumours. Formalin's use as a fixative stems from its capacity for cross-linking of proteins; however, it also forms bridges between protein and nucleic acids, which can cause impurities. Additionally, unbuffered formalin drives acid-mediated hydrolytic nucleic acid fragmentation, which can inhibit PCR amplification.13 DNA extraction methods must therefore preserve nucleic acid integrity while eliminating impurities.
Commercial DNA extraction kits typically combine proteinase K digestion with solid-phase DNA purification. Variations in proteinase enzyme, digestion time and incubation temperature exist between methods.14
DNA quantitation may also impact PCR efficiency.15 Common methods include spectrophotometry, fluorometry and qPCR. Nanodrop spectrophotometry (Thermo Scientific, Waltham, Massachusetts, USA) relies on light absorption by DNA at 260 nm.16 Advantages include ease of use and identifying contamination using absorbance ratios. One disadvantage is that spectrophotometry cannot differentiate DNA/RNA. Qubit Fluorometry (Invitrogen, Carlsbad, California, USA) detects fluorescence from double-stranded DNA-specific dyes but is more laborious. qPCR relies on quantitation from the cycle threshold associated with a template-specific probe. However, qPCR is the most laborious and expensive of these methods.
Although the aforementioned variables have been examined in isolation, their impact on downstream mutation testing is unclear. To assess these variables in practice, we designed a diagnostic RING trial investigating pre-PCR and mutation testing methods of 13 laboratories.
Materials and methods
Experimental design overview
Figure 1 provides a study overview. Thirteen laboratories (table 1) were selected according to their workflow comprising either DNA extraction or BRAF/EGFR mutation testing. Blinded FFPE samples were analysed by participants. DNA extracts were returned to University College London (UCL) for follow-up analysis of purity, integrity and quantitation.
Overview of location, extraction method, quantitation method and mutation detection method for each laboratory

Overview of RING trial design. FFPE, formalin-fixed paraffin-embedded; QFI, quantitative functional index; UCL, University College London.
FFPE sample selection
To assess pre-PCR variation, engineered, cell-line FFPE curls (Horizon Diagnostics, Cambridge, UK) were employed. SW48 and MCF10A cell lines were genetically modified to contain clinically relevant EGFR and BRAF mutations using recombinant adeno-associated virus vectors. The modified cell line was titrated against its matched normal parental cell line to generate mixtures containing quantifiable amounts of EGFR and BRAF mutations. Cells were grown under standard tissue culture procedures, trypsin was used to release the cells and cells were counted using a NC100 Nucleocounter (Chemometec, Denmark). The cells were used to generate FFPE blocks manufactured using a validated, proprietary method to achieve cell numbers of 160 000 cells per FFPE section. Therefore, each FFPE section contained defined theoretical DNA yields (see figure 1 for details). Cell numbers per section were validated theoretically using known core and cell size. Section volume (V=πr2h) was calculated as 15 µm * π * (2500 µm2)=294 375 000 µm3. SW48 and MCF10a cell volume (V=4/3πr3) was calculated as (4/3) * π * (7.5 µm3)=1766 µm3. The potential number of cells comprising each 15 µm section is therefore 166 690 cells. Consequently, the theoretical DNA yield of each 15 µm section is 1.1 µg assuming 6.6 pg DNA content in each cell (theoretical DNA content in diploid cells). This is in accordance with Horizon Diagnostics Aperio (Leica Biosystems, Denmark) cell count data that measured cell numbers across 24 FFPE blocks, demonstrating a mean cell count of 167 681 with an SD of 35 993. Additionally, comparison of DNA yields from 15 µm sections cut from 12 original FFPE blocks was performed by Horizon Diagnostics. The Maxwell DNA extraction kit (Promega, USA) was used to extract DNA from a total of 36 FFPE sections. DNA quantitation was performed using the Quantifluor assay (Promega, USA). The mean DNA yield per section was 465 ng with an SD of 94, which is in keeping with the calculated theoretical yield. The allele ratios of mutations in each sample were quantified using droplet digital PCR (ddPCR) performed with the BioRad QX100 platform (Hercules, California, USA).
Additionally, cell-negative FFPE curls and control FFPE tissue were used as negative and positive extraction controls, respectively. Thus, as shown in figure 1, each participant was sent the following: six FFPE samples to assess pre-PCR steps that included four curls with 1100 ng theoretical DNA yield, one cell-negative curl and one curl comprising FFPE tonsil tissue. Two FFPE samples to assess both pre-PCR steps and mutation detection that included one curl with 1100 ng theoretical DNA yield harbouring 25% BRAF V600E and 33% EGFR G719S, and one curl with 1100 ng theoretical DNA yield harbouring 66% BRAF V600E and 20% EGFR L858R.
Sample preparation
Samples were blinded and labelled 1–8 (figure 1). Laboratories were anonymised (A–M). Samples were shipped at room temperature and stored at 5°C. One sample set was validated at UCL using clinical protocols. DNA was extracted using the QIAamp DNA FFPE tissue kit (Qiagen, Germantown, Maryland, USA). DNA was quantified using Qubit V2.0 (Invitrogen, Carlsbad, USA). BRAF/EGFR screening was performed using Sanger sequencing. Primer sequences are listed in online supplementary table S1. Results were unblinded and confirmed against vendor specifications.
Participant instructions
Participants performed pre-PCR processing of samples 1–8, recorded results and returned remaining DNA from samples 1 to 6 to UCL for follow-up. Participants screened samples 7 and 8 for EGFR/BRAF mutations. Laboratory directors signed the data collection sheets provided.
Follow-up analysis
Follow-up analysis of DNA from samples 1 to 6 (n=78) including quantitation, assessment of integrity and PCR inhibition was performed at UCL and Asuragen.
Quantitation
Repeat DNA measurements (n=78) were performed at UCL and Asuragen using a Qubit fluorometer (Invitrogen). Qubit was selected over qPCR due to its more frequent use alongside Nanodrop.17 The Qubit dsDNA HS Assay Kit (Invitrogen) was used for quantitation. Assay calibration was performed using the internal controls provided. Measurements were correlated between the two sites and compared with participants’ measurements.
DNA integrity
DNA integrity was assessed at UCL and Asuragen using independent methods. Initial analysis was performed at UCL using a Biomed2 multiplex-PCR assay of differently sized amplicons (targeting 400, 300, 200 and 100 bp fragments of AFF1, PLZF, RAG1 and TBXAS1, respectively), which enables visual analysis of DNA fragment size distribution using a 6% polyacrylamide gel.18 ,19 One microlitre of sample DNA was used.
Asuragen assessed DNA integrity using quantitative functional index (QFI)-PCR, which quantifies the DNA templates that are competent for PCR amplification. This method measures the fraction of amplifiable DNA in a sample by determining the copy number of PCR-reportable DNA (referenced to a standard curve of known copies of intact HapMap cell line DNA) divided by the total number of bulk DNA copies (determined by spectrophotometry). As demonstrated by Sah et al20, DNA with low functional quality (and thus a low QFI score) is vulnerable to false negatives and/or false positives in downstream mutation assays.
PCR inhibition
The ‘SPUD’ qPCR assay was employed by Asuragen, which benchmarks a reference Cq value generated from potato-specific DNA against the Cq values generated for 1 µL of the study samples.21 The original method was employed except that 600 copies of the potato-specific DNA target were used to provide more sensitive detection of inhibition.
Statistical analysis
Multivariate statistical analysis including one-way analysis of variance (ANOVA) (not assuming equal variances) was used to assess pre-PCR variation. All statistical analyses were carried out using R V2.14.2.
Results
Participant demographics
The cohort comprised 13 clinical laboratories. Case volume of each laboratory is illustrated in figure 2. Table 1 provides an overview of participant methodology.

Annual number of cases screened by each participant for epidermal growth factor receptor (EGFR) and BRAF mutations demonstrates that the cohort had experience detecting EGFR and BRAF mutations, on average screening >10 and >7 samples per week for EGFR and BRAF mutations, respectively. The volume of our cohort (489) is more than double that of 112 participants in a European external quality assurance scheme, suggesting our findings may be relevant to other laboratories.30
Variation in DNA recovery
The range of DNA recovery from the total number of samples analysed in the ring trial (n=104) is shown in figure 3. Yield variance was highest for (control) sample 6 (mean=4473.1 ng, SD=5508.4 ng, CV=123.1%, n=13) and lowest for sample 2, an engineered sample (mean=1016 ng, SD=1124.7 ng, CV=58.4%, n=13). Significant difference in DNA yields across all samples was seen using ANOVA (p<0.0001); however, variation among engineered samples was not significant (p=0.3782). Consistency among the engineered samples was to be expected due to their artificial nature, demonstrating their suitability for use as reference material. On average, reported yields from engineered samples (n=78) were 1.3-fold greater than the calculated theoretical yield, suggesting potential problems arising during the pre-PCR process (see online supplementary figure S1). Surprisingly, 10/13 laboratories reported yields averaging 235.8 ng (SD=266.7, CV=1.13%, n=13) from cell-negative sample 5.
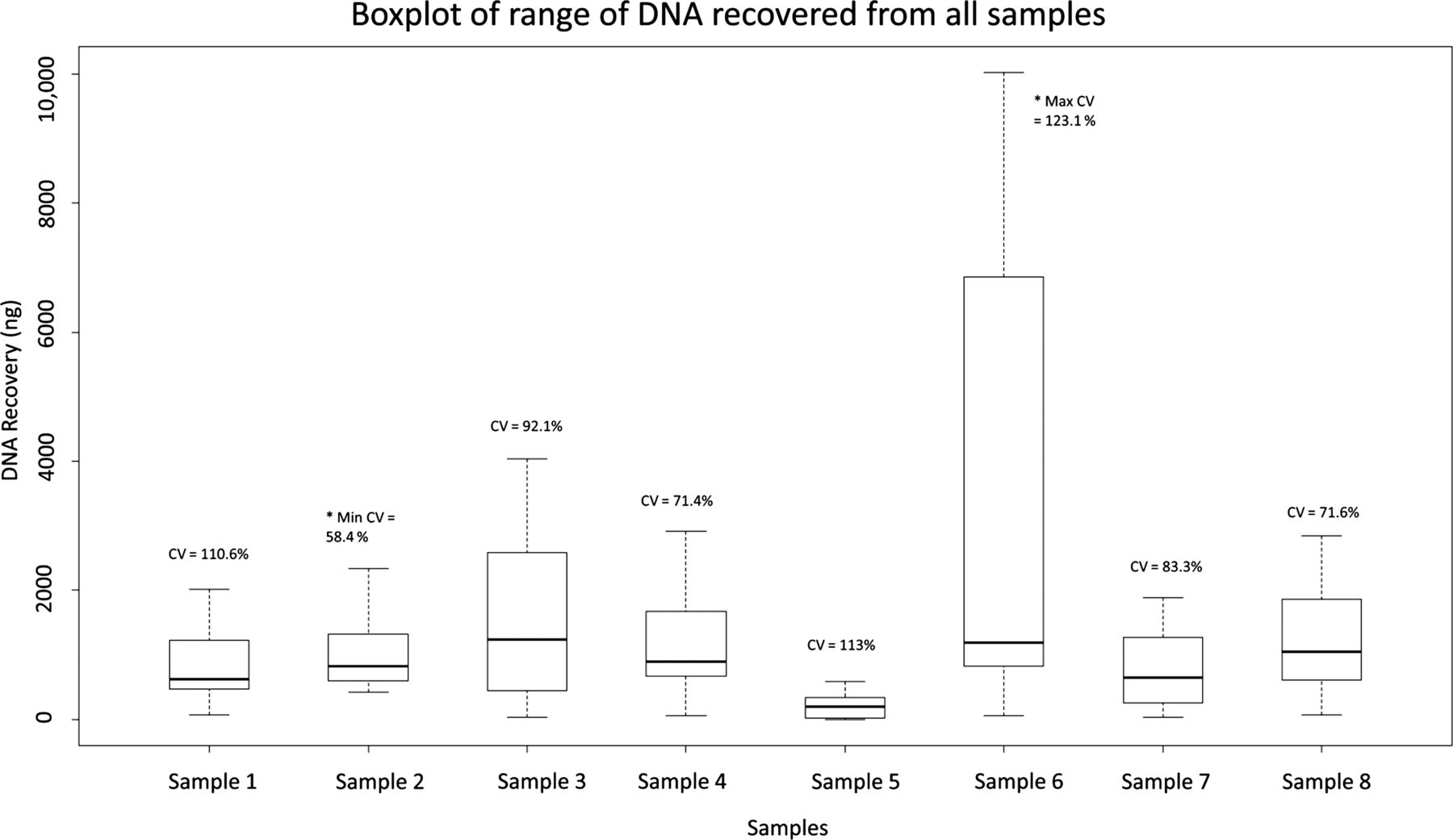
Range of total DNA (in nanograms) recovered from each sample by participants. In total, 104 samples were analysed. Samples 1–4 comprise theoretical DNA yields of approximately 1100 ng, sample 5 comprises a cell-negative curl, sample 6 comprises a tonsil tissue specimen, sample 7 comprises 1100 ng theoretical DNA yield, in turn harbouring BRAF V600E and EGFR G719S, and sample 8 comprises 1100 ng theoretical DNA yield, in turn harbouring BRAF V600E and EGFR L858R. The range of DNA recovered from the engineered samples, after dropping laboratory K outlier values, was >55-fold for two of the six and >5-fold for the four others. Range in DNA recovered from control sample 6, excluding outliers from laboratory K, was >40-fold.
Laboratory variation
Comparing DNA recovery from engineered samples shows laboratory D was most consistent (CV=17.1%) and laboratory K was least consistent (CV=146.7%) (see online supplementary figure S2). Laboratory K failed DNA recovery from 7/8 samples. Laboratory A failed DNA extraction from 2/8 samples.
Mutation analysis
In total, 11.9% (5/42) of potential calls failed to detect the EGFR or BRAF mutations present (table 2). Laboratory K failed to detect mutations in samples 7 and 8 due to insufficient DNA recovery (two calls missed as did not routinely screen EGFR mutations). Laboratory A failed to test sample 7 due to insufficient DNA recovery (two calls missed). Laboratory H extracted adequate DNA but failed to detect 20% EGFR p.L858R in sample 8 using Sanger sequencing (one call missed). This is likely to be due to the limit of detection of Sanger sequencing approaching 20% mutant: wild type.
Overview of mutation detection performance of each laboratory
Variation in pre-PCR steps
Extraction: Quantities of DNA extracted from the engineered samples (measured by Qubit) were compared with respect to the various extraction methodologies employed. The results demonstrate that Qiagen EZ1 had the lowest yield variance (CV=52%), while Qiagen QIAamp had the highest (CV=82%). Despite small sample size, overall yield variance between the methods was relatively low (30%) (figure 4).
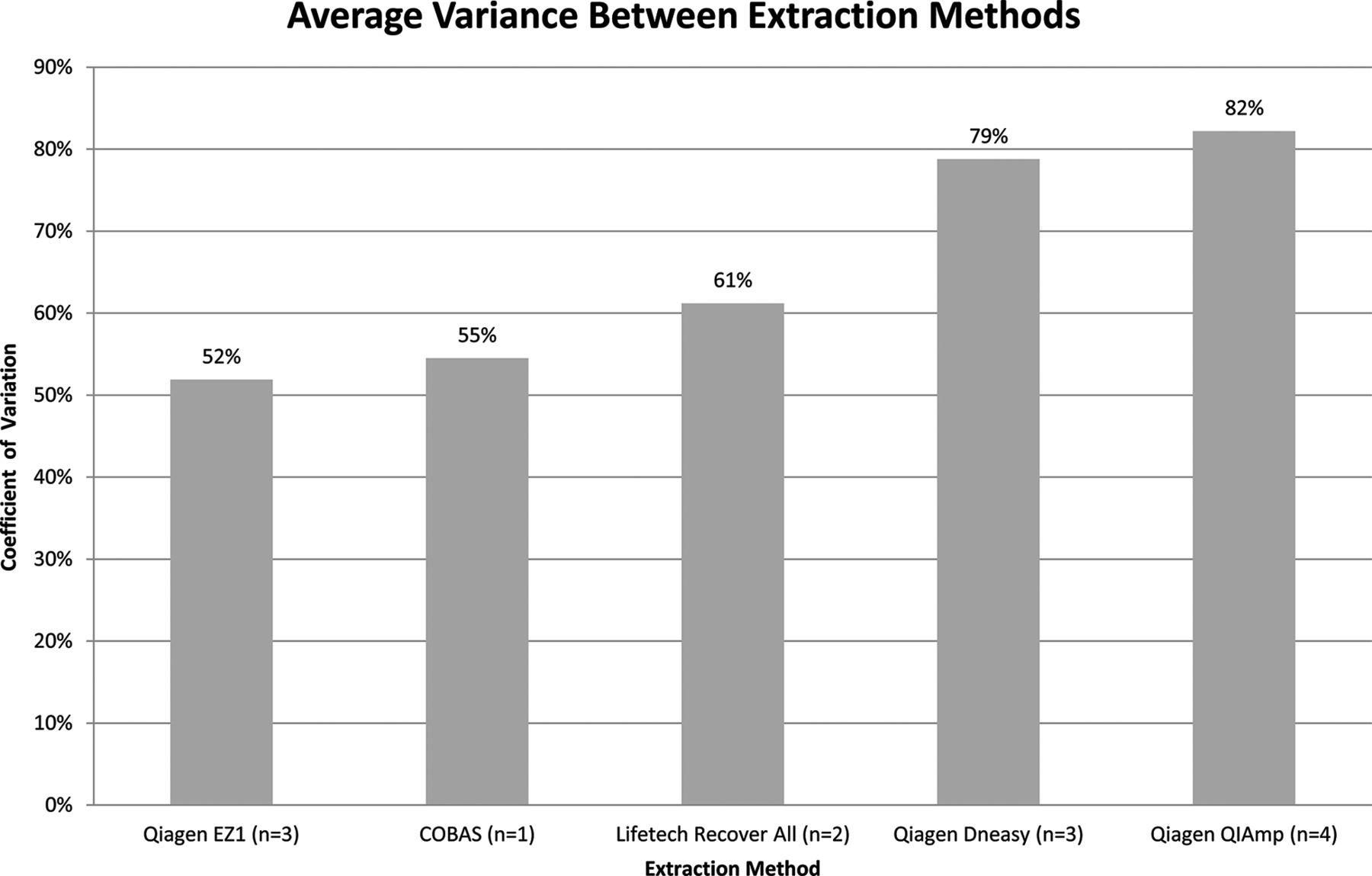
Variance in DNA recovered by the different extraction methods was calculated using Qubit measurements for engineered samples 1–4. It should be noted that one of the laboratories used a modified version of RecoverAll; however, for the purpose of the analysis, these were treated the same. N refers to the number of laboratories using each method.
Quantitation: Correlation between Nanodrop and Qubit measurements for identical samples (n=78) was poor (R2=0.48, p<0.0001) (see online supplementary figure S3). Comparing Qubit versus Nanodrop measurements for identical samples, represented as the Nanodrop:Qubit (N/Q) ratio, demonstrates median Nanodrop readings were 5.1-fold higher than Qubit measurements for the same samples (figure 5). In addition, Qubit measurements of cell-negative sample 5 from all laboratories were undetectable in all cases.

Average Nanodrop:Qubit ratio for each laboratory as well as the average and median ratio for the entire cohort.
DNA integrity: Sample quality (n=78) assessed using multiplex-PCR showed degradation of DNA from control sample 6 (figure 6C). In contrast, DNA from the engineered samples was intact (figure 6A). Analysis of cell-negative sample 5 confirmed absence of amplifiable material (figure 6B), underscoring the significance of DNA recovery reported for these samples. Average N/Q ratio for the degraded control samples (N/Q=15.6) was higher than intact engineered samples (N/Q=10.9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Biomed 2 multiplex PCR results for assessing DNA integrity. (A) Representative set of engineered samples indicates high-quality DNA with four strong bands at 400, 300, 200 and 100 bp. (B) Comparison between engineered samples (B2 and C2) and a representative set of cell-negative samples 5. No bands were seen for cell-negative samples except in samples from laboratories E, I and K. These were not reproducible on repeat. (C) Representative set of control samples from each laboratory shows just one band at 100 bp indicating degraded DNA.
Asuragen assessed samples from a subset of laboratories (D, H, J, L and M) using QFI-PCR, which demonstrated DNA from engineered samples 1–4 to have similar ‘functional’ quality, with approximately 1 in 4–6 input templates amplified.20 In contrast, <1 in 100 templates in DNA from control sample 6 were amplified. These findings have close correlation with the multiplex-PCR data.
PCR inhibition: ‘SPUD’ PCR analysis demonstrates absence of PCR inhibitors in all but one sample. Mean Cq across all samples (Cq=30.21) was consistent with mean Cq from the standard curve for 600 copies of exogenous DNA target (Cq=30.14) in all but one sample, M6 (see online supplementary figure S4).
Discussion
Recent EQA in NSCLC demonstrates suboptimal EGFR testing.22 Since EQA programmes assessing mutation detection from FFPE samples do not usually assess pre-PCR steps, it is difficult to establish causes of error. Although previous studies have assessed individual pre-PCR steps such as DNA extraction from FFPE,23 ,24 accuracy of DNA quantitation15 ,17 ,23 ,25 ,26 ,27 and PCR inhibition,28 ,29 this is the first study, to the best of our knowledge, that has compared both pre-PCR processing and mutation detection from FFPE samples across different laboratories.
In this study, we systematically assessed pre-PCR steps through a RING trial. Reported DNA yields from artificial FFPE samples exceeded the calculated theoretical yields, suggesting the possibility of discrepancies associated with pre-PCR sample processing. The likelihood of this was increased because the majority of participants reported large quantities of DNA from cell-negative samples. Additionally, mutation detection across the cohort was suboptimal. The majority of missed calls (4/5) were caused by insufficient DNA recovery from the engineered samples. Therefore, we discuss some of the important pre-PCR variables and potential solutions laboratories may adopt to reduce variation in their methods.
DNA extraction methods are comparable
Basic criteria for DNA extraction methods include sufficient DNA extraction for analytical testing, removal of contaminants and preservation of DNA integrity. Lack of PCR inhibition, as shown by the SPUD assay, in all but one sample indicates the methods employed by the cohort are good at removing contaminants. Similarly, DNA from the engineered samples remained intact, suggesting the methods preserved DNA quality. Considering that other participants consistently recovered DNA from all engineered samples, it is not clear why laboratories A and K failed DNA extraction. Laboratory K had the lowest throughput (50/year) and may not have had sufficient operator experience. Therefore, operator error may have led to failure to recover DNA from the samples. However, laboratory A, which screens >1000 cases per year, had more experience and yet failed DNA recovery from two samples demonstrating that experience alone was insufficient to guarantee reliable DNA extraction. Overall, comparable DNA quality and quantity (ΔCV=30%) was achieved by the different methods, which suggests that differences in FFPE extraction kit are unlikely to contribute significant variation to the pre-PCR process. However, failed DNA recovery by two laboratories indicates that operator error may be prevalent. Minimum variance associated with the automated Qiagen EZ1 could reflect automated platforms being less prone to operator variability. This is supported by the fact that laboratories employing automated extraction methods (B, C and L) experienced no failures. Therefore, use of automated platforms may reduce operator error. In cases where throughput does not justify automation, use of FFPE extraction controls may help identify instances of DNA extraction failure.
Nanodrop overestimates DNA quantity
DNA quantitation is important because sequencing assays cannot detect low abundance mutations if the minimum number of mutant templates is not available. Almost all participants (12/13) employed DNA quantitation using Nanodrop. This is unsurprising considering Nanodrop's speed and low cost. All laboratories employing Nanodrop, except laboratory E, reported DNA recovery without interpretation of absorbance ratios, which was surprising since they also used DNA extraction methods that co-purify DNA/RNA.
Comparing Nanodrop and Qubit measurements of identical samples (n=78) shows that Nanodrop overestimates DNA fivefold, which could theoretically result in using lower amounts of starting DNA for the downstream sequencing step and cause false negative results. Discrepancies between the two methods were to be expected since Deben et al,25 O'Neill et al27 and Simbolo et al17 reported similar findings. However, Foley et al23 and Haque et al26 provide contradictory reports. Therefore, our evidence further supports discordance between the two. Considering that Nanodrop is not DNA-specific and that laboratories used protocols for DNA/RNA co-purification, discordance due to RNA contamination is likely. However, it does not explain why yields (average=235 ng) were reported for cell-negative samples using Nanodrop. Factors such as DNA fragmentation have been associated with higher Nanodrop readings. For example, Simbolo et al report worse Qubit/Nanodrop correlation for partially degraded versus intact DNA.17 This was confirmed in our study, which showed the N/Q ratio was higher for degraded control samples than intact engineered samples. Deben et al25 investigated RNA extraction from FFPE and found that Nanodrop measurements were 32% higher than Qubit. However, when measuring RNA extracted from non-FFPE material, they found both methods to be similar. Foley et al23 also demonstrated concordance measuring non-FFPE samples. Although these published findings may reflect the effects of formalin-induced fragmentation, we propose that incomplete paraffin removal may also contribute to artificially raised Nanodrop measurements. Although sample contamination was not evident from the SPUD assay results, it is conceivable that the contaminant may only confound ultraviolet light absorbance and have little impact on amplification efficiency. Other factors that may contribute variation to Nanodrop include using less than recommended sample size that can result in incomplete formation of the liquid sample column. Additionally, if the reference/blank solution is not identical to the sample solvent or if the blank sample is not measured first, inaccurate quantitation may occur. However, these factors alone cannot be responsible for the degree of variation observed between Nanodrop and Qubit, nor the discrepancies associated with the blank samples.
Reported DNA yield from cell-negative samples underlines the importance of interpreting Nanodrop results using absorbance ratios. Good quality DNA should have 260/280 nm ratios between 1.7 and 2.0, while 260/280 ratios >2 indicate RNA contamination.26 When using protocols co-purifying DNA/RNA, laboratories should use Qubit or qPCR for DNA quantitation because Nanodrop is not DNA specific. Employing cell-negative controls and commercial gDNA may improve quantitation accuracy. Simbolo et al17 recommend concomitant use of Nanodrop (for measuring sample purity) and Qubit (for accurate DNA quantitation), which offers the most reliable protocol with optimal cost/time requirements.
Measurement of DNA integrity is a useful addition to the pre-PCR process
Extraction methods cannot efficiently purify DNA fragments <100 bp.14 If a given sample does not contain the required number of mutant templates, downstream mutation detection will not be successful. Measuring DNA integrity during pre-PCR may therefore save costs and time. We demonstrated use of QFI-PCR for assessing DNA integrity, which quantifies the percentage of templates competent for amplification.20 Alternatively, the BIOMED2 multiplex-PCR assay enables analysis of the distribution of varying DNA fragment sizes.
Conclusion
In this study, we demonstrate that while mutation detection assays are seen to be reliable, failures in DNA extraction are prevalent. Such failures in DNA recovery represent a waste of precious patient samples, which necessitates repeat testing or even repeat biopsy that can delay critical patient therapy. In addition, we have demonstrated that significant variation exists between DNA quantitation protocols that might cause laboratories to overestimate the quantity of DNA in their samples. Operator error may account for failures observed in manual DNA extraction and may be reduced using automated methods such as Qiagen EZ1, or in manual instances, by incorporating DNA extraction controls. DNA quantitation in conjunction with DNA/RNA co-purification should be performed using Qubit or qPCR since Nanodrop cannot distinguish DNA/RNA. However, Nanodrop absorbance ratios should be used initially to assess contamination. DNA integrity impacts amplification efficiency and should therefore be assessed during pre-PCR. We recommend adaptations to the pre-PCR workflow that may help laboratories reduce variation in their preanalytical process. These adaptations are useful for laboratories carrying out sample preparation for sequencing or for NGS library generation. We also recommend that assessment of pre-PCR processing of FFPE samples be incorporated into EQA programmes. In the design of this RING trial, we have demonstrated a model for robust assessment of key pre-PCR components.
Take home messages
-
Pre-PCR variation is significant among experienced laboratories, while down-stream EGFR and BRAF mutation analysis is shown to be relatively robust.
-
DNA extraction methods used in this study are comparable; however, operator error may have led to failed DNA recovery even in experienced laboratories with high case load.
-
Nanodrop overestimates FFPE DNA compared to Qubit. Qubit or qPCR should be the primary quantitation method, particularly for protocols co-purifying DNA and RNA.
-
External quality assurance programmes should include pre-PCR steps in their assessment of mutation detection from FFPE samples.
Acknowledgments
Special thanks to Surekha Gajree (UCL) for assistance in sample preparation, Keith Miller (UKNEQAS) for facilitating this collaboration, Paul Morrill (Horizon Diagnostics) for providing materials and Tatum Cummins and Hayley Pye for reviewing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
GJL and RAH contributed equally
-
Contributors Conception, design, analysis and interpretation of the work and writing of the manuscript: JRK, GJL and RAH. Data acquisition: TD, JS, MG, IS, LJJ, MML, GJT, DGdC, JAB, AW, JLD, SH, RB and EV.
-
Funding Funding for this research was provided by UCL Division of Surgery & Interventional Science, Horizon Discovery and UCL-Advanced Diagnostics.
-
Competing interests JRK has received an educational grant from Horizon Discovery.
-
Provenance and peer review Not commissioned; externally peer reviewed.