Article Text

Statistics from Altmetric.com
Introduction
Overexpression of the human epidermal growth factor receptor 2 (HER2) protein, mainly due to HER2 gene amplification, in breast cancer is associated with aggressive histological features and poor prognosis.1 ,2 Several randomised clinical trials have demonstrated substantial survival benefits in patients with HER2-positive breast cancer treated with anti-HER2 targeted therapy, such as trastuzumab3–5 and the tyrosine kinase inhibitor lapatinib6–8 but not in HER2-negative patients.9 This, in addition to potential side effects of these costly drugs and evidence of higher response rates to neoadjuvant chemotherapy in HER2-positive tumours,10 has emphasised the need for accurate assessment of HER2 status in patients with all-invasive breast cancer. Early studies, with relatively small numbers of cases, suggested that as many as 30% of breast cancers had HER2 overexpression, with a false positive rate up to 19% and a false negative rate of 5–10%.11–13 However, following publication of guideline recommendations11 ,14–18 and refinement of test performance parameters including the standardisation of tissue handling, assay methodology and adopting quality assurance measures, recent data indicate that the frequency of HER2 positivity is between 13% and 20%.11 ,12 ,19–21 The false positive rate is reduced to less than 6%, the false negative rate is much lower (<2%) and, importantly, the proportion of inconclusive cases is significantly reduced.11 ,12 ,19 ,20 To ensure the highest degree of test accuracy, reproducibility and precision, there is a need to further standardise and improve the quality of technical aspects such as assay performance, validation, proficiency testing and accreditation. These guidelines, which are presented on behalf of the UK National Coordinating Committee for Breast Pathology, aim to update the previous UK guidelines14–16 and provide recommendations on the preanalytical and postanalytical assay performance parameters and give advice on methodology and quality assurance measures for HER2 testing.
Preanalytical measures
Specimens
HER2 status should be assessed in all invasive primary breast carcinomas and in recurrent and metastatic tumours whenever biopsy tissue is available. Bilateral carcinomas, histologically distinct ipsilateral carcinomas or widely separated carcinomas considered to be separate synchronous primary tumours should each be assessed. It is deemed reasonable not to assess multiple ipsilateral tumours if they are histologically similar and colocated in the same quadrant/region of the breast. There is no consensus on testing residual invasive tumour following neoadjuvant therapy, although some recommend this approach. Retesting non-responding stable or progressive HER2-negative tumours particularly high-grade tumours or those with a long time period between preoperative biopsy and excision may be considered but cannot be recommended routinely in view of the lack of evidence.
Excellent concordance between core biopsy and surgical specimens has been shown using immunohistochemistry (IHC) and in situ hybridisation (ISH).20 ,22 ,23 In the majority of UK centres, HER2 testing is performed on the diagnostic needle core biopsy specimens, mainly to ensure timely availability of results at the time of postoperative multidisciplinary team (MDT) treatment planning discussion and also to enable consideration for neoadjuvant treatment use which is increasingly used for operable cases. Although assessment of HER2 status on needle core biopsy is recommended and no repeat on excision specimens is needed if the test is clearly positive or negative, performing/repeating the assay on incisional or excisional surgical specimens should be considered if:
(1) the core biopsy is not available (ie, there is only a cytology sample); or (2) there is a possibility that the HER2 test on the core biopsy is unreliable or unrepresentative of the tumour identified in the resection specimen as follows:
-
HER2 assessment is uninterpretable on the core due to technical artefacts (ie, suboptimal processing or staining) or there is doubt about the core biopsy handling.
-
The core biopsy HER2 status remains in the equivocal category after IHC and ISH; for example, repeat assessment is advised if the core biopsy was scored as 2+ on HER2 IHC with borderline negative ISH (ratio of number of HER2 to chromosome 17 centromere copies of 1.8–1.99 or HER2 gene copy number is 4–6).
-
Invasive tumour on the core is too small for reliable assessment, or if invasive disease is intimately admixed with in situ carcinoma, or only identified in the excision specimen. There is insufficient data to define the amount of invasive tumour tissue in core biopsy sufficient for analysis; however this can be left to the reporting pathologist's discretion.
-
If the tumour in the resection specimens is morphologically distinct from that in the core biopsy, for example of a clearly different histological type or histological grade (eg, low grade on the core and high grade on the excision, but not just reflecting minor difference in the mitotic count or proportion of solid areas).24 A repeat may also be undertaken on concurrent metastatic nodal disease if it is morphologically distinct from the primary breast tumour.
-
If the core biopsy staining is heterogeneous and shows a focus of strong HER2 positivity in <10% of the area of the invasive carcinoma in the core biopsy, HER2 testing should be repeated on the excision specimen. If this pattern is detected on the excision specimen, a different tumour block or a nodal metastasis can be tested, to determine the percentage of positive/amplified tumour present in a larger tumour sample.
Fine needle aspirates from primary breast carcinoma are not suitable for assessment of HER2 status as the distinction between invasive and in situ disease cannot be made on such samples. However, if fine needle aspiration (FNA) is the only material available, or in metastatic disease, some evidence indicates that ISH is reliable for assessing HER2 status in liquid-based and cell block preparations.25 In the case of metastatic bone lesions that require HER2 assessment, it should be noted that decalcification techniques have the potential to influence immunohistochemical assessment in a detrimental manner and such decalcified samples should be tested with ISH methods.26 ,27
Fixation and processing
Good fixation of specimens used for HER2 testing should be ensured and the cold ischaemic time (time from removal from the patient to placing in fixative (cold ischaemic time)) should be as short as possible, certainly less than 1 h.28 Formalin fixed, paraffin embedded tumour tissue samples are appropriate for assay. Tumours samples should be fixed in buffered formalin and embedded in paraffin wax; fixatives containing alcohol can result in staining of normal tissue and use of Bouin's fixative will preclude testing by fluorescence in situ based methods. Other methods of tissue fixation can also adversely affect antigen reactivity. At least 6 h fixation is recommended for core biopsies. Surgical specimens should be incised as soon as possible through the carcinoma to allow initial penetration of fixative and then sliced into 5–10 mm slices to ensure rapid penetration and even fixation. Tissue should be placed in an adequate volume (ideally 10:1; fixative:tissue) of fixative for at least 24 h and not more than 72 h Centres using rapid fixation and processing must validate their methodology for HER2 assessment.
Sections should be stained within 1–2 days of cutting and drying. Excessive section drying time has also been shown to cause a loss of HER2 expression and it is therefore recommended that freshly cut sections are either dried at 60°C for 1 h or 37°C overnight29 (http://www.ukneqasicc.ucl.ac.uk/neqasicc.shtml).
Algorithms for HER2 testing
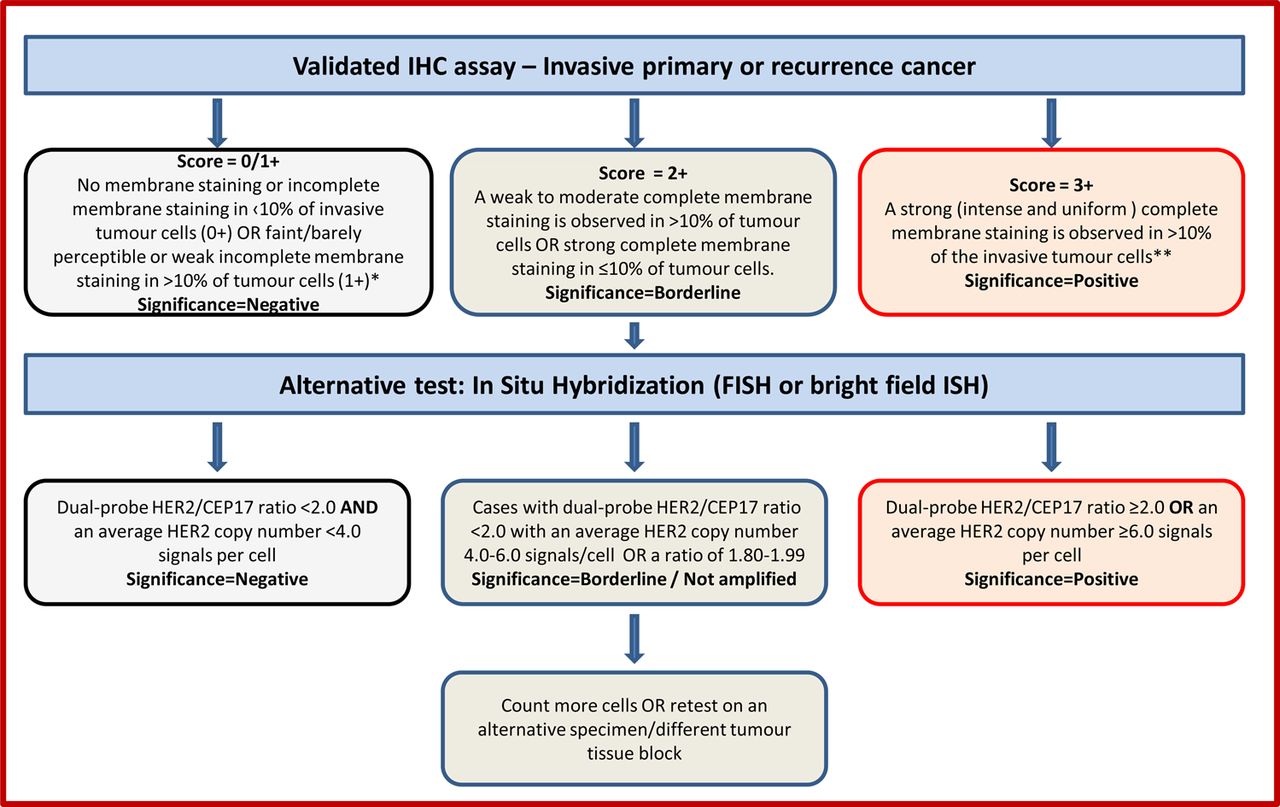
IHC for detection of protein overexpression and ISH for detection of gene amplification status are the techniques recommended for determining HER2 status. High concordance between IHC and gene amplification status is reported.16 ,30 ,31 The current UK recommendations for HER2 testing are for a two-tier system using IHC with reflex ISH testing if required, using the model shown in figure 1, or a one-tier ISH strategy. In general testing is performed using IHC with analysis of equivocal cases by ISH, but this does not preclude laboratories from using primary HER2 ISH testing, particularly if the quality of tissue fixation is questionable.32 ISH has usually been conducted using a fluorescence ISH (FISH) technique. Bright-field ISH, which can be used to assess HER2 status with a regular light microscope, is now accepted as an alternative to FISH.33 The most common bright field ISH uses a DNA probe coupled to a chromogenic ISH or silver ISH detection system, or a combination of both.33 ISH can be conducted using a single probe to enumerate HER2 copies per nucleus or as a dual-probe technique which allows determination of the HER2:CEP17 ratio and HER2 gene copy number. For this reason the inclusion of a chromosome 17 probe is strongly advocated. Currently, other available HER2 testing techniques (PCR, ELISA, Southern blotting, mRNA assays and DNA microarray) should be used for research only. Similarly, HER2 results obtained from a non-ISH technique as part of a prognostic panel cannot be regarded as diagnostic and should not replace standard assay methods detailed above.

Recommended HER2 scoring algorithm for immunohistochemistry (IHC) and in situ hybridisation (ISH).
*Insufficient data is available to comment on moderate complete membrane staining in ≤10% of tumour cells or strong incomplete membrane staining in >10% of tumour cells. A repeat on another specimen/tissue block is advisable.
**Membrane staining must be intense and uniform and resemble chicken-wire. Ignore incomplete or pale membrane staining in the percentage estimation.
Scoring immunohistochemistry
Only membrane staining of the invasive tumour should be considered when scoring HER2. Cytoplasmic staining and staining of in situ disease should not be scored, and normal epithelium should be negative. The HER2 IHC scoring method is a semiquantitative system based on the intensity of reaction product and percentage of membrane positive cells, giving a score range of 0–3+ (figure 1). Samples scoring 3+ are regarded as unequivocally positive, and those scoring 0/1+ as negative. Borderline scores (2+) are regarded as equivocal and mandate further assessment using ISH (figure 1). Appropriate controls featuring different scores (3+, 2+ and 1+/0) should be included in every test run. Some centres also include an on-slide positive control section. The HER2 test should be reported as indeterminate, and repeated where possible, if technical issues prevent one or both tests (IHC and ISH) from being reported as positive, negative or equivocal. Examples include, inadequate specimen handling, artefacts (eg, crush or marked edge artefacts) that make interpretation difficult, analytical testing failure or if controls are not as expected (ie, sample shows strong membrane staining of normal breast tissue). In such a case, an alternative test, or another specimen if available, should be used to determine HER2 status. These guidelines revert to the previously used IHC criterion of >10% cells staining for HER214 ,15 instead of the >30% cut-off used in the previous guidelines.11 ,16
Scoring ISH
HER2 ISH testing, which uses a dual probe method, is initially expressed as the ratio of HER2 signal to chromosome 17 centromeric enumeration probe (CEP) signal. Subsequently the average HER2 gene copy number reporting has been used in some countries when using dual probe and single HER2 gene probe methodology. The UK recommendation is to use dual probe ISH and report the HER2/CEP17 signal ratio and HER2 copy number. Tumours showing a ratio ≥2.0 and/or a mean HER2 gene copy number ≥6 are considered to be positive. Assigning cases as positive based on a HER2 gene copy number ≥6 where the HER2/CEP17 ratio is <2 remains controversial but is recommended as its inclusion aligns with national guidance in other countries.21 Cases with dual-probe HER2/CEP17 ratio <2.0 with an average HER2 copy number <4.0 signals/cells are considered as HER2 negative (figure 1). Classification of cases with monosomy of chromosome 17 and a HER2/CEP17 ratio >2.0 remains controversial with uncertain anti-HER2 treatment benefit34 ,35 but current consensus21 is to regard such cases as amplified.
A selection of normal cells should be assessed to confirm successful hybridisation, detection and visualisation, before assessment of the invasive carcinoma. The number of chromosome 17 and HER2 signals is scored and recorded and the mean HER2 to chromosome 17 copy ratio is calculated for 20–60 cells, where possible using at least three distinct tumour fields. In most cases, where either clear amplification is observed or the ratio is below 1.5, scoring of 20 tumour cells is sufficient. Only cells in which the nuclear borders can be identified should be counted. Overdigested, damaged and truncated nuclei should not be scored. Only cells with minimum one copy of HER2 and CEP17 should be scored. The location of the areas assessed should be recorded. In cases where either tumour heterogeneity is seen, or if the ratio is close to 2.0 or if the average copy number is between ≥4.0 and <6.0 signals/cell, more cells should be scored (at least 60), for details see below. Samples with >2.0 copies of HER2 for each chromosome 17 in the fields assessed are considered to be amplified. The HER2 ISH test should be reported as indeterminate and a repeat/alternative test (on the same or another specimen) is requested in the following situations: Controls are not as expected, nuclear resolution is poor, if a significant proportion of signals are unscorable due to weak signals or >10% of signals occur over cytoplasm, autofluorescence is strong or the observer cannot find and count at least two areas of invasive tumour.
HER2 heterogeneity
Genomic heterogeneity refers to the coexistence of more than one population of tumour cells with distinct HER2 amplification characteristics within the same tumour. Intratumoral heterogeneity can be seen as a clustered form where distinct populations/clones of amplified and non-amplified tumour cells coexist, or as a mosaic form which includes the presence of isolated amplified cells in a predominantly non-amplified tumour or a diffuse mixture of amplified and non-amplified cells across the tumour.21 ,36 While such heterogeneity is generally uncommon in breast cancer, the following approach has been proposed to manage heterogeneous HER2 gene amplification in breast cancer and is recommended in these guidelines37:
In all cases where ISH is performed the entire slide should be scanned before counting, areas of apparent heterogeneity should be identified during this scan and/or by reference to an IHC stained slide. The number of chromosome 17 (CEP17) and HER2 signals should be counted in 20–60 non-overlapping invasive cancer cell nuclei, using at least three distinct tumour fields. If there is evidence of heterogeneity between fields (or less frequently within fields) additional cells (at least 20 per field) and/or fields (up to 6) should be counted. The HER2/CEP17 ratio should be calculated for each field individually. Where the mean HER2/CEP17 ratio in any field is 2.00 or greater, the tumour should be regarded as amplified. For all cases where the ratio is between 1.80 and 2.20 results should be based on counting at least 60 tumour cells, and in cases where heterogeneity is suspected this should be 60 cells per assessed field. In rare cases where amplified and non-amplified tumour cells are intermingled in a single field, interpretation is difficult and evidence is lacking. We suggest that for such cases only the presence of clearly amplified cells, with multiple HER2 signals, is considered evidence of heterogeneity, again evidence is lacking in this area. Current evidence does not support using the existence of small numbers of apparently amplified cells within an individual tumour field to identify heterogeneous amplification.36 ,38
In borderline cases, that is, those with a HER2/CEP17 ratio of 1.80–2.20, additional cells should be counted when possible (optimally a minimum of 60 per case), ideally this should include a dual count (from a second observer; either internally or in a second centre). The optimal approach to improving accuracy in this range is to increase the number of cells counted to 60–120, and/or repeat the test. A ratio of 1.80–1.99, after counting further cells and/or repeating the test, should be reported as borderline but not amplified and include a clear statement that the carcinoma is regarded as HER2 negative (taking the mean HER2 copy number into consideration (mean <6 copies/cell)). A ratio of ≥2.00 should be reported as amplified, and regarded as HER2 positive. Data on the response of patients to trastuzumab whose cancers fall within the borderline amplified category are not available; a statement to this effect can be included in reports. While these guidelines are sufficient for the majority of cases, there are occasions when difficult cases should be referred to expert centres for guidance.
Variation increases with highly amplified samples, and is not critical where the ratio of HER2/CEP17 exceeds 4. Where possible, count all signals, but if this is not possible, for example if clusters are present, then try to estimate the number of signals. Count doublets as a single signal. Where resources permit, representative images can be captured and archived. Difficult cases should be assessed by a second observer. A minimum of 10% of cases should be double-reported to ensure consistency between observers.
The ISH report should include: the number of cells scored, the average HER2 and CEP17 copy number and the HER2/CEP17 ratio. Unusual features should be noted. For heterogeneous cases all these details should be reported for each subclone. If there is a problem in specimen handling and/or processing (ie, non-adherence to the guidelines), this should be documented in the report.
Impact of heterogeneity of HER2 IHC and ISH
Although a cut-off of >10% of the invasive tumour area using IHC is used to define positivity, cases showing complete intense membrane staining in <10% of tumour cells are seen, albeit rarely, and should be considered in the borderline category. In such cases, a repeat of the HER2 IHC test on another specimen (eg, a different tumour block) should be undertaken, to determine the percentage of positive tumour present. If this repeat IHC assessment shows a similar pattern, ISH should be performed. Variation in immunostaining between the periphery and centre of tumours can be due to a fixation gradient.
Defining HER2 positivity using ISH may be complex in cases with intratumoral heterogeneity (see above for scoring methodology). Such genetic heterogeneity affects a proportion of breast cancers (11–40%36) and is more frequently seen in HER2-positive tumours. Although no clinical data is available to guide on the likely response of genetically heterogeneous tumours harbouring HER2-amplified subclones to trastuzumab, it would be valuable to standardise the definition of genetic heterogeneity to facilitate future study of its clinical relevance.36
Evaluation
For assessment of HER2 IHC and ISH preparations, training and experience in interpretation of histological characteristics of breast tissue is essential. Recognition of different histological tumour types is required. In particular, HER2 status should only be determined on the invasive portion of the tumour, and neither IHC nor ISH should be reported in isolation. If it is difficult to differentiate invasive from in situ disease in the index tumour block submitted for ISH, IHC markers for myoepithelial cells can be used.
Image analysis systems may provide alternatives to manual scoring for HER2 IHC and ISH. However, at present, insufficient evidence is available to recommend their routine use in the diagnostic setting.
Quality assurance measures
Controls
-
The inclusion of controls, ideally including on-slide control(s), and their detailed scrutiny are essential to ensure test accuracy. Controls whose HER2 status has been validated and producing results close to important decision-making points are recommended. Tissue-based controls, from breast cancers, should also be used in all assay runs, ideally showing 3+, 2+ and 1+/0 patterns. Control material should be similarly fixed and processed to the test tissue. Control sections should ideally be cut at the same time as the test material. Long-term storage of precut control sections is strongly discouraged. There is no evidence that storage of blocks leads to deterioration of signal.
-
Cell line preparations containing multiple samples of known HER2 status characterised by FISH and IHC and inclusion of a tumour tissue from IHC 3+ case on each slide are useful as additional controls.
Excessive antigen retrieval should be monitored by evaluating normal breast epithelial cells as an internal control. Should membrane staining be identified in the normal cell population, excessive antigen retrieval may have occurred and retesting of the entire run should be considered. Any such tests should certainly be interpreted with great care; it is reasonable to score a 0 or 1+ tumour as negative, but 2+ or 3+ tumours should have staining repeated. If there is doubt between a 1+/2+ result or a 2+/3+ result, either the IHC should be repeated or amplification status should be assessed using ISH. If membrane staining of normal epithelial cells is seen in a number of cases from the same staining run consideration should be made to repeat staining of the whole run.
-
Crushing and edge artefact, particularly affect core biopsies. ISH, or repeat IHC on the surgical specimen, may be needed. The potential gradient effects of suboptimal fixation, particularly in larger surgical specimens, must also be considered in interpretation of staining.
-
It is essential that assay procedures be standardised so that staining is reliable. As there can be variation between batches of reagents, it is vital that controls are assessed critically for every run. New batches of antibody should also be tested before commencing routine application. Use of standardised operating procedures, including routine use of control materials, is recommended.
Appropriate laboratory assay methods
For IHC and ISH based HER2 testing, comprehensive standardisation of methodology, including monitoring of scoring procedures and the inclusion of validated controls, is mandatory. In the UK, participation and satisfactory performance in the UK National External Quality Assessment Scheme for Immunocytochemistry and In Situ Hybridisation (UK NEQAS ICC & ISH) HER2 IHC and ISH modules is a requirement (http://www.ukneqasicc.ucl.ac.uk).
Standardisation of HER2 IHC staining is best achieved by using a commercial kit/assay. Inhouse ‘home-brew’ (laboratory validated) methods are not recommended but, if used, strict protocols need to be followed, including choice of antibody, antibody dilution and retrieval method, each of which can cause variability in staining results. If a commercial kit/assay is used, it is recommended that laboratories adhere strictly to the kit/assay protocol and scoring methodology. Local modifications of techniques can lead to false positive and negative results. Therefore, it is important to check and audit controls carefully in order to ensure test accuracy. Laboratories using bright field ISH should perform an initial validation against FISH.
Interobserver variation in the assessment of IHC staining can lead to misclassification of HER2 status. Each individual assessor should standardise scoring against known positive, negative and borderline cases. It is also preferable to assess comparability of scoring with a colleague on a regular basis. Before undertaking evaluation of HER2, assessors should receive relevant training.
Published data suggest that interobserver variation is significantly lower for FISH than for IHC. However, especially when developing a new service, care needs to be taken. The recommendation is that laboratories should perform validation studies by dual observer scoring when training new staff until there is concordance of 95%. For ISH validation purposes, each staff member should perform a minimum of 100 ISH tests in parallel with an experienced ISH scorer to attain a minimum concordance of 95% on diagnostic results (amplified and non-amplified status) and numerical results (for HER2 and CEP17). Continued monitoring of scoring offers advantages in quality control and training, but is not a requirement.
Validation of standardised assay methodology
Test conditions should be optimised so that distinct moderate or strong membrane staining shows >90% concordance with HER2 ISH positive samples. This can be achieved by:
-
Dual HER2 IHC and ISH assay of a contemporary series of breast carcinomas (minimum 100 cases). Use of tumour tissue array blocks for this purpose may reduce costs. HER2 ISH assay can be confined to those cases demonstrating 3+, 2+ and 1+ membrane reactivity.
-
Alternatively, a series of carcinomas that have already been scored for HER2 IHC and ISH, from a reference laboratory, can be used.
Laboratories not able to standardise inhouse methodology should also consider using a commercial validated kit assay system.
ISH for HER2 gene evaluation
ISH testing for HER2 should meet the following criteria:
-
Comprehensive standardisation of methodology
-
Validated controls: the inclusion of a chromosome 17 probe to allow for correction of the HER2 signal number for chromosome 17 aneusomy (seen in ∼30% of cases and reportedly more common in tumours that show discrepant HER2 expression and in tumours with discordant HER2-protein and gene copy number measurements) is recommended.
Case load
-
Laboratories providing a testing service should be carrying out a minimum of 250 assays per year for immunohistochemical detection of HER2. This target level has been set to ensure higher consistency of assay quality and continuing expertise of assay providers.
-
Centres with low numbers of cases (<250 per year) should consider using a reference laboratory service.
-
Similar principles apply to ISH testing; it is recommended that laboratories testing < 100 cases per year (<150 including gastric carcinomas) consider referral of their workload to a reference laboratory. A smaller case load has been set for ISH assay, as it is generally accepted to be a more discriminant test at the positive–negative borderline, has greater ease of methodological standardisation, and has less observer variation.
General principles
ISH should be performed on the same block as used for IHC. It is advisable that areas of the invasive carcinoma to be scored with ISH are located using a serial section stained with H&E and HER2 IHC where available. Care should be taken to avoid areas of ductal carcinoma in situ, which can show amplification even when adjacent invasive tumour cells are negative.
Tissue digestion should be standardised to maintain nuclear morphology and should follow strict protocols. Some laboratories find it helpful to evaluate nuclear structure before hybridisation and to adjust digestion, where appropriate, to preserve nuclear integrity. This may be particularly valuable with difficult sections, bone biopsies, etc. Evaluation of sections before hybridisation can also improve efficiency and is recommended. Hybridisation and washing steps should be standardised. Guidance can be provided by the reference laboratories. Use of automated tissue processors and standardised commercial tissue digestion kits can improve consistency and should be considered.
It is recommended that commercially available validated probes are used. There are a number of commercial kits for HER2 ISH using fluorescence and chromogen based detection systems and which are all acceptable, once properly validated.
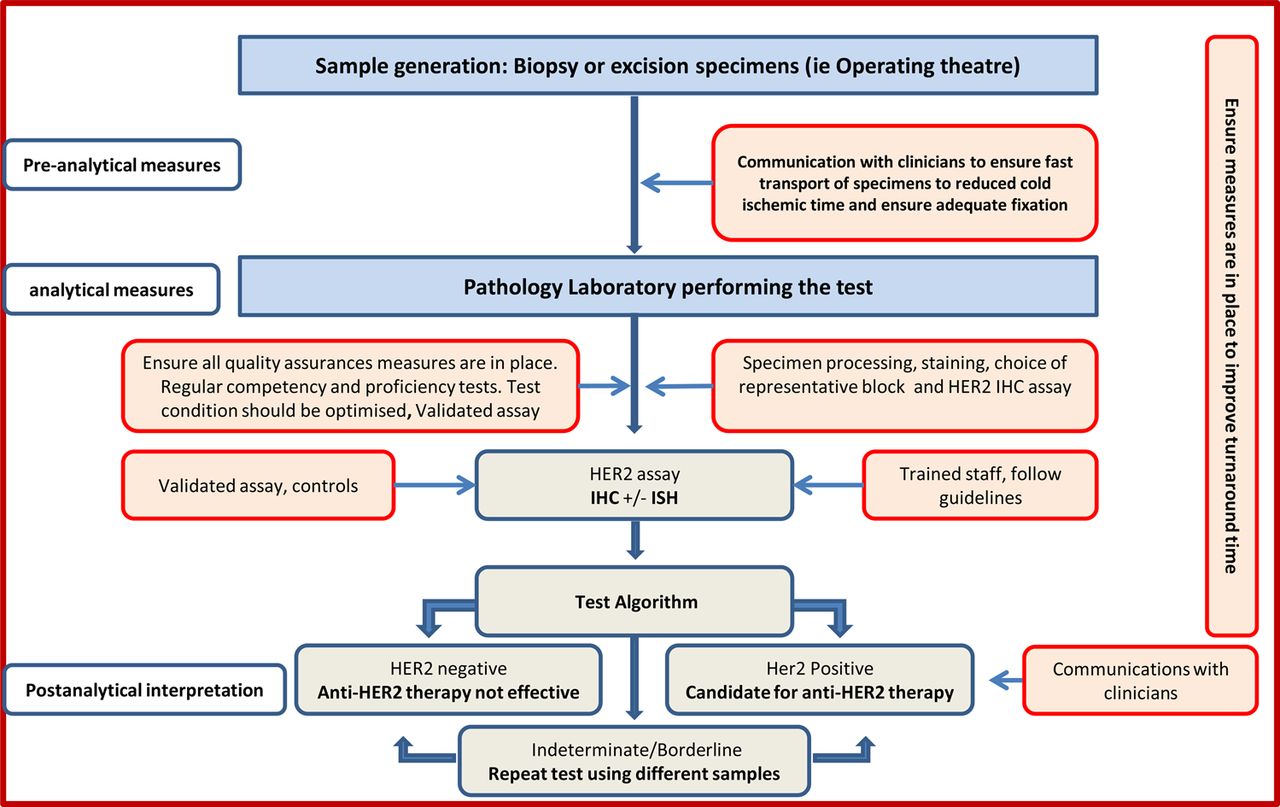
Short turnaround times for HER2 testing that do not delay the management of patients are recommended. Turnaround time is recognised to be variable between different centres, and can be addressed at the level of cancer networks and local services (figure 2). The National Institute for Care Excellence recommends that HER2 status of the tumour be assessed and the results made available within 2 weeks to allow planning of systemic treatment by the MDT and that local arrangements and written clinical protocols are in place to ensure HER2 status results are available within this time (http://publications.nice.org.uk/breast-cancer-quality-standard-qs12/quality-statement-5-pathology-er-and-her2-status#quality-measure-5). It is also important to emphasise the role of improved communication between pathologists/laboratories performing the test and clinicians to ensure proper handling of specimens (ie, prefixation time and fixation type), short turnaround time and proper interpretation of the test results.

{kind=link}
{kind=link}
Pathway for HER2 testing.
Audit
Regular and ongoing audit should be undertaken. Laboratories should audit their overall positive rate for HER2 using a combination of IHC and ISH. It is important to ensure that the sample size is adequate. Of note, the average proportion of invasive breast cancer cases recorded as HER2-positive is 14.5% (UK NEQAS ICC & ISH combined 5 year national audit data), with 14.3% of primary carcinomas and 18% of metastatic cases being HER2-positive (table 1). Of these cases approximately 22% cases are reported as borderline (2+) on IHC; of which 15–16% are reported as HER2 ISH amplified.16 The proportion of HER2-positive breast cancers found in screen detected breast cancer cases is recognised to be lower than in symptomatic practice. Audit of HER2 assay turnaround time is also important as it is critical to patient pathway.
Proportion of HER2-positive primary and metastatic breast cancers*
Quality assurance for HER2 receptor evaluation in the UK
All UK clinical laboratories using IHC or ISH to assess HER2 status as a predictive marker must participate in an appropriate external quality assurance programme, such as that run by the UK NEQAS ICC & ISH.
Communication
In the era of personalised medicine and the commonplace routine practice of MDT meeting for discussion of diagnosis and management of all patients with cancer in the UK, improved communication within the team is considered of paramount importance. Although for many years there has been collaboration between pathologists and patient-facing clinicians in the UK, this guideline further emphasises the importance of this collaboration. Close communication with surgeons and radiologists is therefore advised in order to improve control over samples prefixation time and fixation type, and with oncologists to improve understanding of interpretation of the results. This is also expected to facilitate control over HER2 test turnaround time.
In conclusion, this update contains recommendations supported by a sufficient level of evidence on key points related to HER2 testing methodology, testing algorithm, interpretation of the results and the potential need for retesting. Laboratories offering a HER2 testing service and pathologists, oncologists and surgeons involved in test interpretation and clinical use should do their best to adhere to published guidelines and ensure accurate performance and interpretation of such tests.
References
Footnotes
-
Correction notice A sentence under Scoring ISH section has been corrected since published Online First.
-
Collaborators Dr R Adamson, Dr Al-Sam, Dr M Ashton, Dr N Anderson, Prof G Callagy, Dr S Cawthorne, Dr D Coleman, Dr N S Dallimore, Dr R Deb, Dr D Fish, Dr A Girling, Dr S Hales, Mr K M Horgan, Dr M Howe, Mrs S Kodikara, Mr K Lea, Prof L Jones, Dr G McCusker, Dr E Mallon, Dr D M Parham, Dr N Patel, Prof J Patnick, Dr C M Quinn, Dr D Rowlands, S J Sellars, Prof T J Stephenson, Dr C A Wells and Prof R Wilson.
-
Contributors EAR and IOE prepared the draft. SEP and JMSB contributed to the initial drafting. All coauthors contributed to the final version of the manuscript and approved it.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.