Article Text

Abstract
Chronic neutrophilic leukaemia (CNL) is a rare type of myeloproliferative neoplasm (MPN) characterised by sustained leucocytosis (≥25×109/L) with neoplastic proliferation of neutrophilic granulocytes in blood and bone marrow. In contrast to chronic myeloid leukaemia, the disease primarily involves neutrophilic lineage with persistent proliferation of mature forms of neutrophils. No consistent cytogenetic changes have been reported. Known recurrent genetic changes in other MPNs such as JAK2, MPL, CALR, BCR-ABL1, PDGFRA, PDGFRB and FGFR1 are mostly absent. Recently, mutations in colony stimulating factor 3 receptor (CSF3R) have been reported in high frequency in CNL. This discovery has provided more insight into its pathogenesis and opened up possible treatment options. In this article, we review the clinical findings, morphology, pathobiology and differential diagnosis of CNL and treatment implications of CSF3R mutations.
- MYELOPROLIFERATIVE DISEASE
- HEMATOPATHOLOGY
- HAEMATO-ONCOLOGY
Statistics from Altmetric.com
Introduction
The classification of myeloproliferative neoplasms (MPNs) and definition of chronic neutrophilic leukaemia (CNL) have undergone several changes over the past century. Tuohy reported the first case of CNL in 1920 in a female patient with massive splenomegaly and neutrophilic leucocytosis.1 The neoplastic nature of CNL could not be clearly established at that time. Subsequent studies reported CNL cases as a variant of chronic myeloid leukaemia (CML).2 With accumulation of similar cases, it became more evident that these cases were distinct from CML and would be better categorised as a subgroup in myeloproliferative disorders.3 ,4 The French-American-British (FAB) classification of myeloid neoplasms did not recognise CNL as a distinct entity mainly due to its rarity.5 Later, the 2001 WHO classification recognised CNL as chronic myeloproliferative disease and published the diagnostic criteria, which were basically a diagnosis of exclusion.6 The recent 2008 WHO classification updated the diagnostic criteria with inclusion of additional criteria, specifically the exclusion of gene rearrangement of PDGFRA, PDGFRB or FGFR1.7 To date, the diagnostic criteria for CNL have been based on a lack of knowledge of specific morphological and genetic markers. Recently, however, oncogenic mutations have been reported in the majority of the patients with CNL.8 Availability of these markers will likely change our diagnostic approach to CNL in the near future.
Epidemiology
Since first described in 1920 by Tuohy, approximately 200 cases of CNL have been reported to date.7 The literature includes many case reports and a handful of small case series. The diagnostic criteria for CNL have only been defined relatively recently; it is unclear from the literature how many are true cases of CNL. It is likely that less than 40% of reported cases meet the current WHO diagnostic criteria.9
With rare exceptions, CNL is primarily a disease of elderly with most patients presenting in their 60 s. The sex distribution is nearly equal. A review of 33 published cases of CNL reported a median age of 62.5 years and male to female ratio of 2:1.10
Clinical features
Many patients are asymptomatic at the time of diagnosis. In other patients, fatigue is the most common symptom.9 A small number of patients present with weight loss, night sweats, bone pain, gout or pruritus.11 The most common and consistent finding on physical examination is splenomegaly. Hepatomegaly can be seen,12 ,13 but lymphadenopathy is uncommon.14 Some cases in the literature have reported increased incidence of haemorrhagic diathesis and mucosal bleeding.11 ,12 The bleeding tendency could result from thrombocytopenia and platelet dysfunction or from vascular wall infiltration by the neoplastic leucocytes.15 ,16
Pathology
Peripheral blood
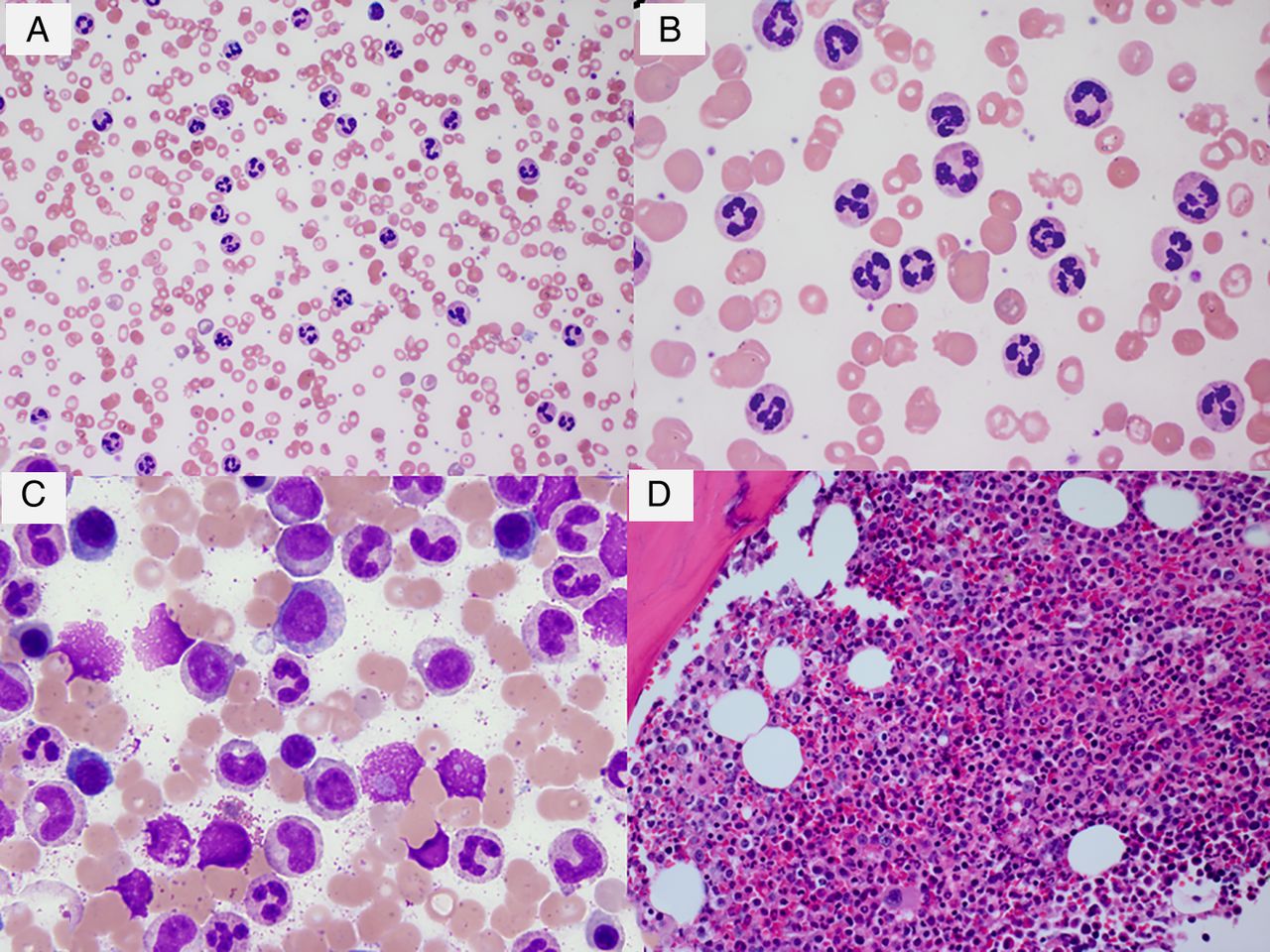
Peripheral blood shows leucocytosis with neutrophilia. The white cell count is moderately elevated with an average of 50×109/L.10 The diagnostic leucocyte count threshold in the current WHO 2008 classification is ≥25×109/L.7 In contrast to CML, leucocytosis in CNL consists of a proliferation of primarily mature neutrophilic granulocytes (figure 1A, B). Segmented neutrophils and bands comprise >80% of the total white cells.7 Neutrophils frequently show toxic granulations and Döhle bodies, which suggests an activated state.17 Features of dysplasia are usually absent. There is no increase in monocytes. Myelocytes, metamyelocytes, myeloblasts and nucleated red cells are rare. In particular, circulating myeloblasts are almost never seen. Platelet count is usually normal except in late stages when mild thrombocytopenia can be seen with increasing splenomegaly and progression of disease. Vitamin B12 levels are elevated due to release of transcobalamin from the granulocytes and precursors.11 As with other myeloid malignancies, serum uric acid levels are elevated due to proliferation of the haematopoietic precursors.11 Leucocyte alkaline phosphatase score is normal or elevated but this is usually a non-specific finding.11 ,13

(A) Peripheral blood smear showing leucocytosis with neutrophilia without left shift (Wright–Giemsa stain, 500× original magnification). (B) High power view of the peripheral blood smear (Wright–Giemsa stain, original magnification, 500×). (C) Bone marrow aspirate smear showing maturing granulopoiesis without increase in blasts (Wright–Giemsa stain, original magnification 1000×). (D) Bone marrow biopsy with marked myeloid hyperplasia. The megakaryocytes are normal in morphology (H&E stain, original magnification 400×). BM, bone marrow; CNL, chronic neutrophilic leukaemia; PB, peripheral blood.
Bone marrow
The bone marrow is hypercellular with marked myeloid hyperplasia. Myeloid to erythroid ratio is frequently more than 20:1. There is an increase in myelocytes, metamyelocytes and bands, but blasts or promyelocytes are not increased typically (figure 1C, D). Unlike CML, eosinophilia or basophilia are absent. Erythroid precursors are relatively reduced and show normal maturation. In general, megakaryocytes are normal in number and morphology. Some cases may show mild megakaryocytic hyperplasia.7 Significant dyspoiesis is not seen; if present, should prompt one to rule out atypical CML (aCML). Reticulin fibrosis is not observed.
Rare cases of CNL have been reported to harbour clonal plasma cells in bone marrow. Bone marrow should be carefully examined for plasma cells, and appropriate immunohistochemical stains should be performed while diagnosing CNL.18 ,19 In cases where clonal plasma cells are detected, the diagnosis of CNL should be supported by molecular or cytogenetic studies to prove clonality.7
Extramedullary tissues
Splenomegaly is a consistent finding in CNL. Infiltration of spleen by CNL primarily involves red pulp cords and sinuses. The white pulp is relatively spared from leukemic infiltration. The red pulp is expanded and filled with segmented neutrophils and precursors. Rarely, megakaryocytes and erythroid precursors can be seen along with neutrophils.13 Similar to spleen, involvement of the liver is seen as infiltration of the sinuses and portal areas by neutrophils.11 ,12 Lymph node involvement has been reported only rarely.14 ,20
Cytogenetics
Many case reports and a few small series have supported the clonal nature of CNL based on X-inactivation partners, karyotypic abnormalities and human androgen receptor gene assays.21 In majority of the cases, cytogenetic studies showed a normal karyotype.7 One review series reported cytogenetic abnormalities in 37% cases of CNL.9 The reported abnormalities included trisomy 8,22 trisomy 21,11 deletion 11q23 and deletion 20q.24 The most frequent cytogenetic abnormality was deletion 20q.9 20q deletions are not specific for CNL and have been reported in other MPNs. It is possible that the reported cytogenetic abnormalities are secondary events in pathogenesis and represent cytogenetic evolution.25
Molecular genetics
The major breakthrough in the pathogenesis of CNL came in 2013 when mutations in the gene coding for colony stimulating factor 3 receptor (CSF3R) were reported in 16 of 27 patients (59%) with CNL or BCR-ABL1 negative aCML. The reported mutation frequencies were 89% in CNL cases (8/9) and 40% in aCML cases (8/18).8 These findings were supported by another study that reported 100% frequency of CSF3R mutation in 12 patients with WHO-defined CNL.26
The CSF3R gene maps on chromosome 1p34.3 and encodes the transmembrane receptor for granulocyte colony stimulating factor (G-CSF; CSF3). It is known to play an important role in proliferation and differentiation of granulocytes.
Two types of mutations are found in CSF3R: the majority occurs in the extracellular domain (membrane proximal point mutations) and a small number occurs in the cytoplasmic portion of the receptor (non-sense or frameshift mutations) leading to truncation of the cytoplasmic domain. The most common membrane proximal mutations include T618I and T615A. These mutations result in ligand-independent activation of CSF3R that initiates downstream signalling through JAK2.8 The point mutation is usually present in isolation or can be seen along with compound frameshift or non-sense mutations. CSF3R mutations have not been reported in patients with reactive neutrophilia.8 ,26
Since the initial discovery of the JAK2 V617F mutation, a few cases of CNL with this mutation have been published.27 ,28 Although rare, when detected JAK2 V617F mutation indicates the clonal nature of the disease. Other mutations that have been reported to occur in CNL include SETBP1,29 calreticulin (CALR)30 ,31 and ASXL132 mutations. In one study, SETBP1 mutations were seen along with CSF3R T618I mutations in 33% patients.26 A recent study has reported SETBP1 and ASXL1 mutations in 14 cases of CNL with mutated CSF3R. Eight cases (57%) showed SETBP1 mutation while five (38%) cases showed ASXL1 and/or SETBP1 mutations. Presence of coexisting SETBP1 and CSF3R mutations may indicate a worse prognosis.32 Two of the cases in this study with coexisting CSF3R and ASXL1 mutations progressed to chronic myelomonocytic leukaemia.
Differential diagnosis
Correct diagnosis of CNL requires careful exclusion of many close mimickers of CNL. Neutrophilia can be associated with a variety of reactive causes including infections, inflammatory disorders and malignancies. The main differential diagnoses of CNL include reactive causes, aCML, plasma cell dyscrasia associated neutrophilia and neutrophilic CML (CML-N). A diagnostic algorithm is depicted in figure 2.
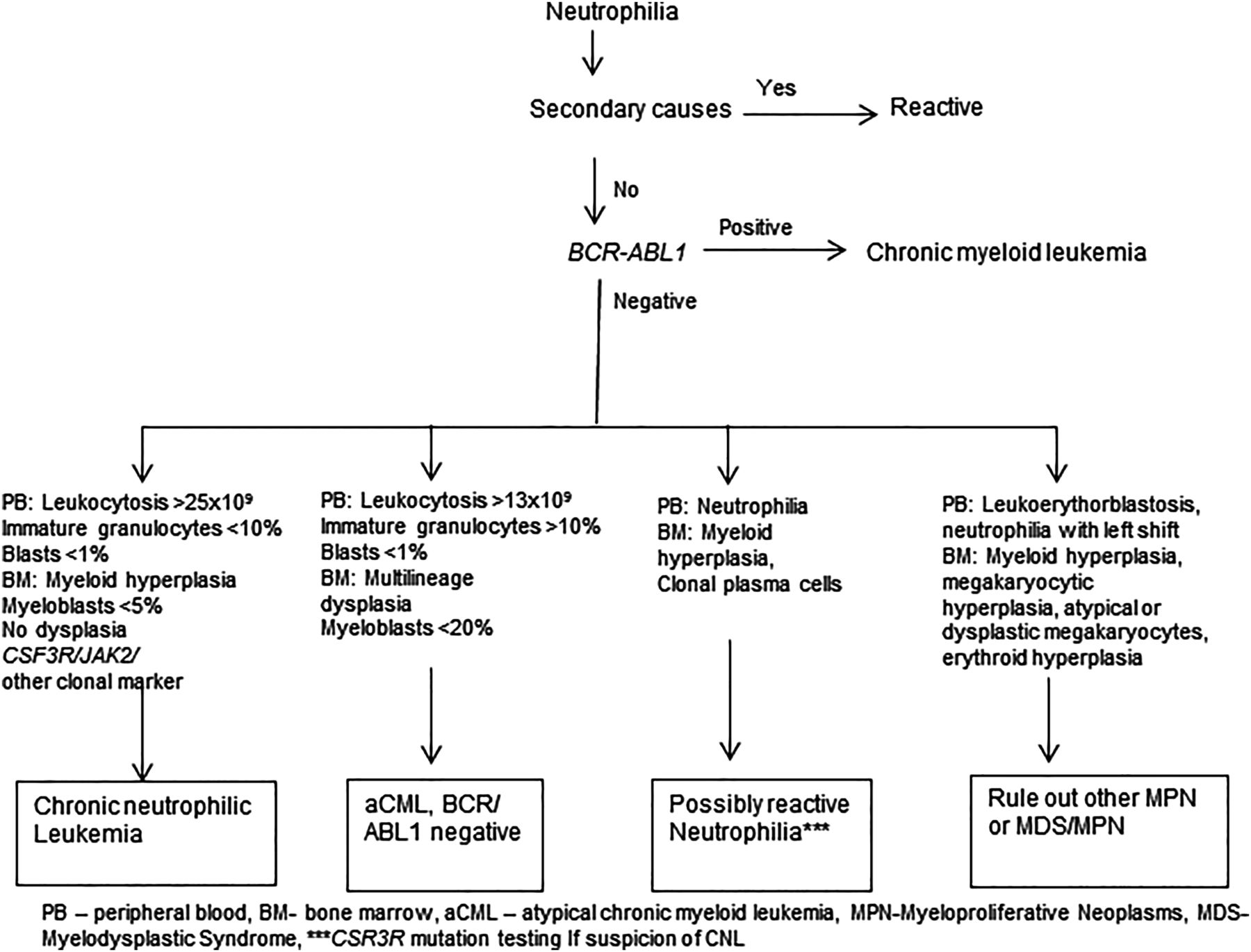
{kind=link}
{kind=link}
Flow chart diagram for diagnosis of chronic neutrophilic leukaemia.
Leukemoid reaction
Leukemoid reaction is the most common cause of neutrophilia. A detailed clinical history is useful to rule out underlying chronic infection or malignancy. The neutrophilia associated with malignancies could result from several factors, such as bone marrow metastasis, inflammatory reaction to necrosis, or production of cytokines by the tumour or in response to it.33 The white cell count is modestly elevated; however, rare cases with white cell count up to 100×109/L have been reported.34 Several cytokines are reported to play role including granulocyte-monocyte CSF (GM-CSF), interleukin-1 (IL-1), G-CSF and interleukin-6 (IL-6). The possibility of underlying occult malignancy should be excluded in all cases of sustained and unexplained neutrophilia. A careful clinical history, a thorough clinical evaluation and a period of observation are usually helpful to rule out these reactive causes. Detection of CSF3R mutation can be of great value in cases with unexplained persistent neutrophilia.
Neutrophilic-chronic myeloid leukaemia
CML-N is a less aggressive variant of CML and closely resembles CNL.35 It is characterised by t(9;22) with the presence of p230 BCR/ABL fusion gene. Three types of fusion transcripts are observed with t(9;22); the p190 and p210 are more common, and the p230 is rare. The shorter transcript p190 is present in a fraction of precursors B-cell lymphoblastic leukaemia, and the slightly longer transcript, p210, is seen in all cases of CML. The p230 fusion transcript is least common.36 Progenitor cells expressing the p230 oncoprotein retain the capacity to progress through the normal stages of granulocytic differentiation and show neutrophilia.37 Many of these cases were classified as CNL. On retrospective analysis, majority of the reported cases of CNL with p230 showed features similar to classic CML rather than CNL, such as thrombocytosis and early blast transformation.38 ,39 There is consensus in the literature now that all cases with p230 should be regarded as CML-N.7
Neutrophilia associated with plasma cell dyscrasia
Plasma cell neoplasms are reported to occur in CNL in up to 32% of the cases, an incidence that is difficult to explain by mere coincidence.40 One explanation is that both processes originate from a common progenitor cell origin that differentiates into myeloid and plasma cells. However, no common clonal markers have been reported in these cases.40 Findings from several studies have supported the idea that neutrophilia is a reactive phenomenon and is driven by an altered cytokine milieu in plasma cell dyscrasia.40 ,41 Elevated IL-1 levels can induce secretion of G-CSF from monocytes, bone marrow stromal cells and endothelial cells.40 Some studies have established the polyclonal nature of neutrophils in these conditions.42 One study demonstrated reduction in G-CSF levels and peripheral blood neutrophil count in a patient with sustained neutrophilia and plasma cell dyscrasia after pulsed steroid therapy.43 While majority of the studies support that the neutrophilia associated with plasma cell dyscrasia is reactive process, cases of CNL coexisting with plasma cell neoplasms may indeed exist. Two cases of JAK2 V617 F positive CNL and coexisting monoclonal gammopathy have been reported recently.26 ,44 However, the question still remains if CNL and plasma cell myeloma are clonally related.
Atypical chronic myeloid leukaemia
aCML belongs to the group of myelodysplastic/MPNs and is characterised by neutrophilic leucocytosis associated with dysplasia. The white cell count is usually elevated. Circulating blasts comprise less than 5% and are always less than 20% of all white cells.6 Granulocytic dysplasia is usually a consistent finding in aCML. Neutrophils show hypolobation, bizarre nuclear segmentation and abnormally clumped nuclear chromatin. The bone marrow is hypercellular and shows features of multilineage dysplasia.45 If optimal peripheral blood and bone marrow aspirate smears are available, aCML can be easily ruled out. Ancillary studies are not often helpful due to substantial overlap in cytogenetic and molecular markers.
Other myeloid neoplasms
Before making a diagnosis of CNL, the possibility of other common MPNs—namely primary myelofibrosis (PMF), polycythaemia vera (PV) and essential thrombocythaemia—should be considered. Primary PMF in prefibrotic phase shows leucocytosis and may show little alteration in red cell morphology. It is crucial to evaluate the morphology of megakaryocytes in the bone marrow biopsy to rule out PMF that shows clusters of atypical megakaryocytes.46 Rare cases of PV have been reported that showed transition to CNL or a similar disease.47–49 Some earlier studies have reported cases of CNL associated with dysplasia or evolving from myelodysplastic syndrome (MDS).50 By current definition, any dysplasia excludes the diagnosis of CNL. Disorders with effective granulopoiesis as well as myelodysplasia are best classified under the broad category of MDS/MPN.
Clinical course and disease progression
The clinical course of the disease is variable. The survival time ranges from 6 months to more than 20 years.10 ,12 Unlike CML, there are no established criteria for progression. The disease progression is characterised by progressive neutrophilia, worsening splenomegaly, resistance to previously effective therapy, anaemia and thrombocytopenia. Transformation to acute myeloid leukaemia (AML) has been reported in 10%–15% of cases.10 A review of 40 cases of CNL reported median overall survival of 23.5 months and median time to progression to AML of 21 months.25 The most frequently reported causes of death were intracranial haemorrhage, progressive disease and treatment-related toxicity from chemotherapy or transplantation.24 A recent study evaluated role of various factors for prognostication including age, lactate dehydrogenase levels, splenomegaly, haemoglobin level, thrombocytopenia, total bilirubin levels, SETBP1 mutation, ASXL1 mutation and ‘T618I versus other CSF3R mutation’ in a group of 14 cases of CSF3R mutated CNL. On a multivariate analysis, only ASXL1 mutation and thrombocytopenia were found to be independently predictive of short survival. The median survival in this group was 23.2 months.32 A trend of short survival has been reported in patients with coexisting CSF3R and SETBP1 mutations.26 A case of CNL has been reported with coexistent CSF3R and SETBP1 mutations that showed in vitro lack of response to JAK inhibitor.29 A study reported transformation of two SETBP1 mutated cases of CNL to AML. The same study also reported evolution of CNL to chronic myelomonocytic leukaemia in patients with presence of ASXL1 mutation and lack of SETBP1 mutation.32 These cooperative mutations likely play an important role in disease transformation.
Future directions
Based on the recent discovery of the CSF3R mutation, Tefferi et al have proposed revised diagnostic criteria for CNL.31 These include: (1) peripheral blood leucocytosis of ≥13×109/L, (2) peripheral blood neutrophils and bands >80%, (3) presence of CSF3R T618I or other membrane proximal CSF3R mutations. The authors also suggest inclusion of 10 other minor diagnostic criteria that are quite similar to current diagnostic criteria. According to these revised criteria, diagnosis of CNL requires the presence of all three major criteria or the presence of the first two major criteria and all minor criteria. The suggested lowered threshold for white cell counts (13×109/L) represents a level above 3 SDs, which is also currently one of the diagnostic criteria for diagnosis of aCML.45
The two types of CSF3R mutations may have different implications in terms of sensitivity to class of tyrosine kinase inhibitors. Reportedly, CNL cases with truncation mutations exhibit in vitro sensitivity to SRC family-TNK2 inhibitors (dasatinib), and cases with membrane proximal mutations are sensitive to JAK inhibitors (ruxolitinib).7 Ruxolitinib has been reported to be effective in a mouse model of CNL and a patient with CNL and a CSF3R T618I mutation.50 These findings anticipate further investigations in the near future into the role of JAK and other tyrosine kinase inhibitors in CNL. The role of additional subclonal mutations such as CALR, JAK2, SETP1 and ASXL1 is being evaluated. SETBP1 and ASXL1 are emerging as new prognostic indicators while awaiting more conclusive studies.
Summary
CNL is a rare MPN that is characterised by persistent neutrophilic leucocytosis in peripheral blood and bone marrow and by frequent hepatosplenomegaly. With the recent discovery of CSF3R mutations as a consistent finding in CNL, it is expected that the current WHO classification will be revised to incorporate this marker in the diagnostic criteria in the near future. Before testing for CSF3R mutations, one should always keep in mind that CNL is a very rare myeloid neoplasm. While molecular testing of CSF3R mutations will quickly become available in diagnostic laboratories, other common causes of neutrophilia must be ruled out before considering the confirmative molecular test.
Take home messages
CNL is a rare myeloproliferative neoplasm which is characterized by persistent peripheral blood neutrophilia.
CSF3R mutation has been recently reported in CNL and is present in virtually all cases.
Role of co-operative mutations such as SETBP1 and ASXL1 mutations in disease progression is being explored.
Before making a diagnosis of CNL and ordering molecular testing, reactive causes of neutrophilia must be ruled out.
Acknowledgments
The authors thank Jennifer Wilson, Writing Center, Thomas Jefferson University, for proofreading and improving the composition of the manuscript.
References
Footnotes
Handling editor Mary Frances McMullin
Contributors The concept of this review was created by GU. GU prepared first draft of the manuscript. JG is responsible for critical revision. All the authors have given final approval for the submission.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.