Article Text

Abstract
Aims Myeloproliferative neoplasms (MPN) are a heterogeneous group of clonal proliferative bone marrow diseases characterised by extensive megakaryocytic hyperplasia and morphological atypia. Despite knowledge of genomic defects, the pathobiological processes driving these megakaryocytic abnormalities in MPN remain poorly explained. We have explored the proliferative, apoptotic and epigenetic profiles of megakaryocytes in human MPN.
Methods Immunohistochemical staining was performed on bone marrow trephine biopsies of 81 MPN (with and without JAK2V617F and CALR mutations) and 15 normal controls to assess the megakaryocytic expression of biomarkers associated with proliferation (Ki67), apoptosis (Bcl-XL, BNIP-3) and epigenetic regulation (EZH2, SUZ12).
Results Myeloproliferative megakaryocytes showed significantly greater expression of proliferative Ki67 and anti-apoptotic Bcl-XL, reduced pro-apoptotic BNIP-3 and increased SUZ12 compared with controls. In essential thrombocythaemia, large-giant megakaryocytes with hyperlobated nuclei showed a trend towards a proliferative signature. In contrast, myelofibrotic megakaryocytes with condensed nuclear chromatin, and cases with CALR mutations, had significant reductions in pro-apoptotic BNIP-3.
Conclusions Uncontrolled megakaryocytic expansion in MPN results from a combination of increased proliferation, attenuated apoptosis and defective epigenetic regulation with CALR mutations favouring apoptotic failure. The higher platelet counts reported to be seen in MPN with CALR mutations may be due to greater dysregulation of megakaryocyte apoptosis.
- BONE MARROW
- PROLIFERATION
- APOPTOSIS
- fibrosis
Statistics from Altmetric.com
Introduction
The Philadelphia negative myeloproliferative neoplasms (MPN) are a group of clonal disorders characterised by proliferation of one or more haemopoietic lineages.1–3 The classification of the MPN entities, essential thrombocythaemia (ET), polycythaemia vera (PV) and primary myelofibrosis (PMF), is guided by the WHO.1–5 Diagnostic criteria include clinical features, bone marrow histopathology and JAK2 mutation status.1–5 All MPN are characterised by megakaryocytic hyperplasia with varying morphologies.5–8 In PV the mega-karyocytes are markedly pleomorphic, and tend to form loose clusters.5–7 In ET megakaryocytic hyperplasia is the major finding with characteristic large and giant forms showing nuclear hyperlobation.5–7 The megakaryocytes in PMF are most abnormal: they cluster in large sheets adjacent to trabecular bone and exhibit pleomorphic morphologies with large hypolobated nuclei. Pyknotic and bare megakaryocyte nuclei are also characteristic.5–7
MPN are characterised by genomic aberrations including driver JAK2V617F (up to 98% of PV; 60–70% ET and PMF),9–12 CALR (20–30% ET and PMF)13–16 and MPLW515K/L (5–8% ET and PMF) mutations.17–19 Less commonly JAK2 exon 12 (approximately 2% of PV)20–22 and other mutations (eg, EZH2, SUZ12, LNK, CBL, SOCS1)23 ,24 may be present. The Janus activated kinase/signal transducers and activators of transcription (JAK-STAT) pathway is a major signalling cascade implicated in MPN.23 ,24 The gain-of-function, missense JAK2V617F mutation promotes constitutive JAK-STAT signalling, extensive cellular hyperplasia and abrogated apoptosis.25–27 CALR mutations (most commonly frameshift lesions involving 52 bp-deletions and 5 bp-insertions) target the calreticulin gene (CALR).13–16 Calreticulin is a calcium-binding chaperone protein that is involved in protein folding, endoplasmic reticulum calcium metabolism, fibroblast proliferation and apoptosis via caspase-8 activation and anti-apoptotic protein cleavage.28 The pathological relevance of these CALR lesions are poorly understood in MPN but correlate with a clinical phenotype of younger age, higher platelet counts, leucopenia, reduced thrombo-haemorrhagic risk and improved overall survival.13–16 ,29
Despite knowledge of these driver mutations, the downstream pathological biomarkers contributing to the megakaryocytic abnormalities in MPN remain unclear. Data from in vitro studies indicate that there is enhanced megakaryocytic proliferation.30 Specifically, CD34-positive cells from patients with PMF cultured in the presence of thrombopoietin and stem cell factor generate 24-fold more megakaryocytes than normal CD34-positive cells.30 Even though the cell cycle biomarker Ki67 is widely applied as an indicator of proliferative capacity in other haemopoietic malignancies (eg, diffuse large B-cell lymphoma, mantle cell lymphoma), it has not been used to assess megakaryocyte proliferation in primary material of human MPN.31 ,32
The JAK-STAT and calreticulin signalling cascades directly participate in regulating cell survival.25 ,28 Human and murine studies have shown that the B-cell lymphoma 2 (Bcl-2) family of apoptotic effectors are perturbed in MPN.33–36 Anti-apoptotic Bcl-XL (B-cell lymphoma extra-large), which prevents apoptosis by limiting pro-death biomarkers Bak and Bax, and the subsequent release of cytochrome c from mitochondria,34 ,35 ,37–39 has been reported to be overexpressed in ET and PMF.30 ,40–43 Bcl-XL expression is also important for platelet production. Deletion of Bcl-XL and MCL-1 has been linked with Bak-Bax mediated mitochondrial apoptosis and depletion of normal megakaryopoiesis. This leads to morphological atypia and thrombocytopenia through reduced platelet shedding.37–39 ,44 In vitro work has shown that thrombopoietin-driven JAK-STAT signalling preserves Bcl-XL and Mcl-1 levels to maintain megakaryoblast survival and platelet manufacture.44 In contrast, BNIP-3 (Bcl-2/adenovirus interacting protein 3) promotes apoptosis by opening the mitochondrial permeability transition pore under hypoxic conditions45–47 and directly inhibits Bcl-XL.36 ,48 Data relating to BNIP-3 expression in myeloproliferative megakaryocytes are however conflicting; some groups report reduced BNIP-342 ,49 whereas others have shown upregulation.50 There is also evidence that calreticulin is defective in myeloproliferative megakaryocytes with CALR mutations.51 Although these studies present some conflicting data, they have led to hypotheses that the megakaryocytes in ET have an enhanced anti-apoptotic profile,41 ,44 whereas in PMF megakaryocytic hyperplasia results from a combination of increased generation and decreased apoptosis.30 ,49
These proliferative and apoptotic megakaryocytic ‘signatures’ may also be influenced by alterations in epigenetic regulators.23 ,24 Enhancer of Zeste 2 (EZH2) and Suppressor of Zeste 12 (SUZ12) epigenetic regulators, molecules that together form the Polycomb Repressive Complex 2 (PRC2) are mutated in up to 10% of MPN.52–55 EZH2 drives the lysine-specific histone methyltransferase (HMT) activity of the PRC2 to repress target genes via the targeted trimethylation of lysine-27 of histone-3.56–60 SUZ12 assists the conformational folding of EZH2 to attain functional HMT activity and tethers the PRC2 to the target nucleosome.56 ,61 Little is known about the PRC2 in normal myelopoiesis but abnormally upregulated EZH2 and SUZ12 have been reported in haemopoietic malignancies (eg, chronic myeloid leukaemia, mantle cell lymphoma, Hodgkin's lymphoma).58 ,62 ,63 In these neoplasms it is thought that PRC2 overexpression confers proliferative and survival advantages to the malignant cells.58 ,62 ,63 A murine model has also shown that mice which overexpress EZH2 develop MPN.64 To our knowledge, neither EZH2 nor SUZ12 have been explored for their effect on myeloproliferative megakaryocytes and their potential contribution to the megakaryocyte pathology.
Although previous studies have explored the mechanisms underlying the megakaryocytic hyperplasia and morphological atypia in MPN, the causative factors remain poorly defined and are likely to be multifactorial. In the present study we have studied primary human bone marrow samples from patients with MPN with and without JAK2V617F and CALR driver mutations. We have focused on proliferation, apoptotic pathways and epigenetic regulators in MPN. Protein biomarkers have been assessed using antibodies to formalin-mercury resistant epitopes in bone marrow biopsies using an immunohistochemical approach and light microscopy assessment.
Materials and methods
Bone marrow samples
Archived formalin-mercury fixed, acid decalcified, paraffin-embedded bone marrow trephine biopsies (n=96) collected between 2000 and 2014 were obtained from patients attending PathWest Laboratory Medicine (Western Australia, Australia). Patients had provided consent for their bone marrow to be studied. All cases were reviewed (WNE and SLH) and classified according to MPN entity (table 1) in accordance with WHO morphological criteria.5 The study included 39 cases of ET, 37 of myelofibrosis (MF) (including PMF and post-ET/PV fibrosis), 5 of PV and 15 normal bone marrow controls. Tissue microarrays (TMAs) of the bone marrow trephine were generated using the TMA Master tissue microarray (3DHistech, Australia). Cores of 1.5 mm were derived from representative areas of each biopsy and each had 50–150 megakaryocytes (as determined through CD61 immunohistochemical staining). JAK2V617F mutation testing was performed by allele-specific PCR, and CALR mutations were determined in accordance with the methods outlined by Nangalia et al.14
Summary of the cases studied
Immunohistochemical staining
TMAs were sectioned at 4 μm onto charged glass slides (Hurst Scientific, Australia). Monoclonal antibodies were to formalin-mercury resistant epitopes and were validated on control tissue prior to their application on human MPN trephine. Antibodies used were CD61 (clone 2f2, Leica Biosystems, Australia), cell cycle-associated Ki67 (clone MIB-1, Dako, Australia), Bcl-XL (clone 54H6, Cell Signalling Technologies, USA), BNIP-3 (clone ANa40, Abcam, UK), EZH2 (clone 6A10, Leica Biosystems) and SUZ12 (clone 3D10, Thermo Scientific Pierce Antibodies, Australia). CD61 was included in all experiments as a megakaryocyte positive control. Immunohistochemical staining was performed on an automated Leica BOND RX immunostainer (Leica Biosystems) after heat induced epitope retrieval with BOND ER1 (Leica Biosystems; pH ∼6) or ER2 solutions (Leica Biosystems; pH ∼9) as per manufacturer's instructions. An alkaline phosphatase BOND Polymer Refine Red Detection kit (Leica Biosystems) was used to detect antigen binding. Sections were counterstained with Mayer's haematoxylin and cover-slipped for analysis by light microscopy. Positive and negative megakaryocytes were counted by a minimum of three observers and the per cent positive calculated for each antigen. Megakaryocyte morphology (ie, size, nuclear configuration) and staining intensity for each antigen were visually and qualitatively assessed. Tissue areas and/or cells of interest were photographed at varying magnifications using a Pixera Pro 600ES microscope camera (Pixera Corporation, USA).
Statistical analysis
Mean megakaryocyte percentage positivity and SD were calculated according to MPN subtype and JAK2V617F/CALR mutational status for each marker. A one-way Kruskal-Wallis ANOVA with post hoc Dunn's tests and Mann-Whitney U tests were performed to evaluate the significance (p<0.05) of megakaryocyte positivity in relation to subtype, JAK2V617F and CALR mutation status. All statistical analyses were conducted using GraphPad Prism V.6.03 software (GraphPad Software, USA).
Results
MPN versus controls
All antibodies used gave the expected reactions with no background staining evident in any haemopoietic cells. Positively stained megakaryocytes were easily identified based on the combination of morphology, red chromogen and nuclear haematoxylin stain. There were significant differences in the per cent positive megakaryocytes expressing Ki67, apoptotic-associated Bcl-XL and BNIP-3, and the epigenetic regulator SUZ12 between MPN and normal bone marrows (figure 1). MPN had approximately 2.7-times greater number Ki67-positive megakaryocytes than normal controls, U(84)=115.5, p<0.0001. Ki67 expression was primarily nuclear and strong with some megakaryocytes having weaker punctate nuclear positivity (figure 2A, B). A small number of megakaryocytes in MPN showed cytoplasmic Ki67 positivity, due to disruption of the nuclear membrane seen during the M-phase of endomitosis (figure 2A). Assessment of apoptotic pathways showed MPN to have twice as many Bcl-XL positive megakaryocytes than controls, U(76)=62.5, p<0.0001 (figure 1). The expression intensity of Bcl-XL tended to increase with megakaryocyte size, as determined by light microscopy appearance and visual morphological interpretation (figure 2C). There were significantly fewer pro-apoptotic BNIP-3 positive megakaryocytes in MPN than the controls, U(72)=31.0, p<0.0001 (figures 1 and 2D). Analysis of the epigenetic regulators showed significantly more SUZ12-positive megakaryocytes in MPN than controls, U(81)=201.5, p=0.0005, but no difference for EZH2 (figure 1). There was no association between EZH2 or SUZ12 positivity and megakaryocyte morphology (figure 2E, F).

Percentage of megakaryocytes positive for Ki67, Bcl-XL, BNIP-3, EZH2 and SUZ12 for all myeloproliferative neoplasm (MPN) and control cases. MPN cases showed a significantly greater percentage of Ki67, Bcl-XL and SUZ12 positive megakaryocytes compared with controls. Mean BNIP-3 was lower in MPN than controls and there was no difference for EZH2. Statistically significant difference, p<0.001 (***).

Representative images of megakaryocyte expression using an alkaline phosphatase chromogen for Ki67 in (A) essential thrombocythaemia (ET) (600×) showing megakaryocytes with nuclear and cytoplasmic positivity, and (B) myelofibrosis (MF) (600×). (C) Bcl-XL positive megakaryocytes in MF (400×) and (D) BNIP-3 negative megakaryocytes in MF (1000×). Epigenetic regulators (E) SUZ12 showing the majority to be positive (600×) and (F) EZH2 nuclear expression in ET (600×).
Essential thrombocythaemia versus Myelofibrosis
Biomarker expression was correlated with morphological entity, namely, ET and MF. PV was excluded from this analysis due to insufficient cases for comparative statistical analyses. Normal controls were also excluded. There were differences between ET and MF megakaryocytes for BNIP-3, SUZ12 and Ki67 (figure 3). MF had significantly fewer BNIP-3 and SUZ12 positive megakaryocytes than ET, with U(57)=248.5, p=0.0082, and U(64)=337.0, p=0.0120, respectively. In MF small megakaryocytes with pyknotic nuclear chromatin were generally BNIP-3 negative (figure 2D). The mean Ki67 mega-karyocyte positivity in MF was less than in ET, but this was not statistically significant, U(66)=418.5, p=0.0759. In ET, Ki67 was expressed in megakaryocytes of all sizes and the stain intensity was unrelated to nuclear lobulation (figure 2A). No differences were seen for megakaryocytic Bcl-XL and EZH2 between ET and MF.

Percentage of megakaryocytes expressing Ki67, Bcl-XL, BNIP-3, EZH2 and SUZ12 in essential thrombocythaemia (ET) and myelofibrosis (MF). (A) Mean BNIP-3 megakaryocyte positivity in MF was significantly reduced compared with ET (p=0.0082). The proportion of SUZ12 expressing megakaryocytes in ET was significantly greater than MF (p=0.0120). Statistically significant difference, p<0.05 (*) and p<0.01 (**). (B) Model illustrating the progressive loss of SUZ12 and BNIP-3 as megakaryocytes progress from ET to MF.
JAK2V617F and CALR mutation status
Megakaryocyte positivity was analysed by mutation status, that is, JAK2V617F+(n=36), CALRMut (n=13) and ‘double negative’ (ie, DN:JAK2V617F−/CALRWT; n=20). A Kruskal-Wallis ANOVA revealed that BNIP-3 megakaryocyte positivity in relation to mutation status was significantly different, H(2,34)=10.90, p=0.0043. The major finding was that CALRMut cases had significantly fewer BNIP-3 positive megakaryocytes than JAK2V617F+, Dunn's MRD(30)=12.79, p<0.01 (figure 4). Morphological review showed no difference in megakaryocytic number between CALRMut and JAK2V617F+ or DN. There was a tendency for CALRMut cases to have greater numbers of mega-karyocytes with a high nuclear:cytoplasmic ratio and pyknotic chromatin pattern. There was a non-significant trend for JAK2V617F+ and CALRMut cases to have fewer EZH2 and SUZ12 positive megakaryocytes than DN cases, Dunn's test p>0.05 (figure 4). There were no significant differences between these genetic groups for Ki67 or Bcl-XL (data not shown).
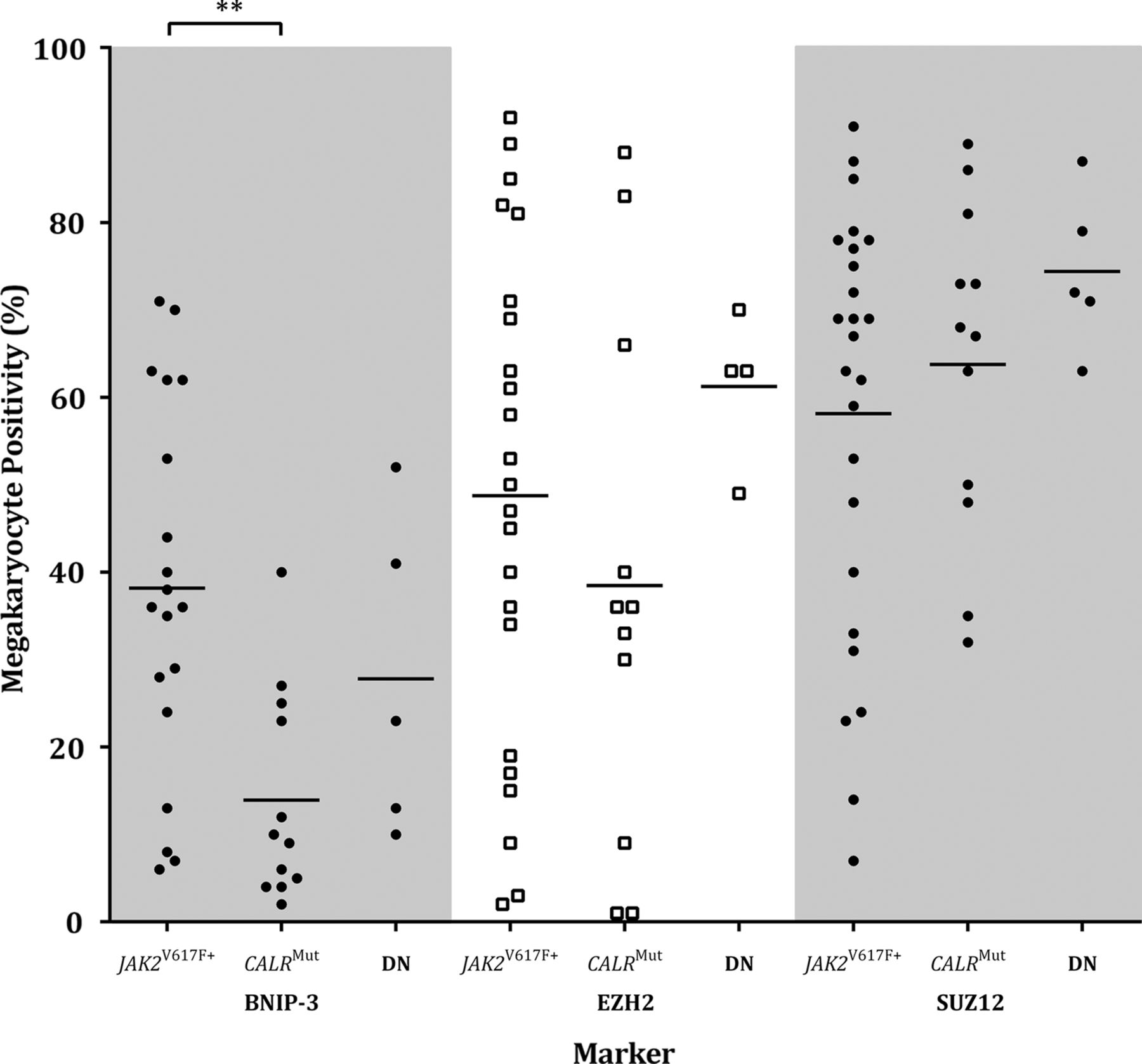
BNIP-3, EZH2 and SUZ12 megakaryocyte positivity for myeloproliferative neoplasm cases by JAK2V617F and CALR mutation status. Mean BNIP-3 megakaryocyte positivity was significantly lower in CALRMut cases compared with JAK2V617F+ (p<0.01). The proportion of EZH2 and SUZ12 expressing megakaryocytes was reduced in JAK2V617F+ and CALRMut compared with JAK2V617F−/CALRWT cases. Statistically significant difference, p<0.01 (**). DN, double negative (ie, JAK2V617F−/CALRWT).
Discussion
Despite improvements in our understanding of the genetic drivers of MPN (eg, JAK2V617F, CALR mutations), the pathological processes underpinning megakaryocytic hyperplasia and morphological atypia in MPN entities remain poorly defined.5–7 ,9–16 Here we demonstrate that myeloproliferative megakaryocytes have enhanced proliferative capacities, reduced apoptotic functions and dysregulated epigenetic control (figure 5). Together, these abnormalities promote uncontrolled megakaryocytic expansion in MPN. We report that MPN cases with CALR mutations specifically showed defective apoptosis (ie, reduced BNIP-3). Since normal megakaryocyte apoptosis is required for platelet production, this dysregulation may explain the higher platelet counts previously reported in clinical studies to be associated with this driver mutation. 13–16 ,29 The precise mechanism for this greater thrombocytosis than in JAK2V617F may be a combination of megakaryocyte expansion and defective platelet production. Differences in ‘signature’ were seen between ET and MF: increased megakaryocytic proliferation, as evidenced by Ki67 expression, was the major aberration in ET, whereas in MF the megakaryocytes had enhanced survival compounded by disrupted epigenetic regulation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagram illustrating that megakaryocytic hyperplasia in myeloproliferative neoplasm is due to a combination of hyperproliferative signals, enhanced cell survival and disturbed epigenetic regulation. JAK-STAT signalling activates the transcription of proliferative (ie, Ki67) and apoptotic (ie, Bcl-XL) genes. In response to Ki67 accumulation, SUZ12 and EZH2 genes are transcribed to form the PRC2 which counteractively represses proliferation promoting genes. There is upregulated SUZ12, but not EZH2, in myeloproliferative megakaryocytes and especially among ET. Anti-apoptotic Bcl-XL is increased in myeloproliferative megakaryocytes to inhibit cytochrome c release and prevent apoptosis. Altered calreticulin signalling through CALR mutations disrupts its ability to correctly fold BNIP-3 to attain pro-apoptotic functionality. Reduced BNIP-3 availability, especially in MF megakaryocytes, decreases its ability to mitigate the anti-apoptotic effects of Bcl-XL. Bcl-XL, B cell lymphoma extra-large; BNIP-3, Bcl-2/adenovirus interacting protein 3; Calr, calreticulin; Casp-8, caspase-8; Cyt-c, cytochrome c; EED, embryonic ectoderm development; ET, essential thrombocythaemia; EZH2, enhancer of Zeste 2; GF, growth factor; JAK2, Janus activated kinase 2; MF, myelofibrosis; P, phosphate; R, receptor; SUZ12, suppressor of Zeste 12; STAT, signal transducers and activators of transcription.
Megakaryocytes are actively proliferating cells that undergo multiple rounds of endomitosis, and in MPN, can generate up to 512N.8 We show that in MPN there are 2.7-times more Ki67-expressing megakaryocytes in active phases of the cell cycle compared with controls, thereby promoting megakaryocytic hyperplasia. The Ki67 expression is predominantly nuclear, which is indicative of continuous endomitotic preparation and cycling.65 There is also a trend towards more Ki67-positive megakaryocytes in ET than MF, suggesting that hyperproliferative Ki67 signals drive megakaryopoiesis in ET. Further, Ki67 was expressed in ET megakaryocytes of all sizes, including characteristic large and giant forms with hyperlobated nuclei (figure 2A). This implies that the deregulated proliferative advantage persists throughout megakaryopoiesis and promotes continual megakaryocyte expansion, even in large polyploid megakaryocytes where it is expected that these cells would have reached their proliferative threshold.8
Myeloproliferative megakaryocytes also showed enhanced survival capabilities as a consequence of disruptions to anti-apoptotic and pro-apoptotic modulators. In keeping with findings from other studies,40–42 ,49 we detected significantly increased anti-apoptotic Bcl-XL in neoplastic megakaryocytes. They also had significant reductions in pro-apoptotic BNIP-3 which would further constrain the capacity of the mega-karyocyte to mitigate Bcl-XL survival signals and block Bak-Bax mediated mitochondrial apoptosis.36 ,45–48 The most pronounced reductions in BNIP-3 were in MF, suggesting a loss of pro-apoptotic potential during progression to the ‘accelerated’ phase of MPN,1–3 ,66 as well as those with CALRMut. In this study we also have morphological confirmation that small megakaryocytes with condensed nuclear chromatin (characteristic of MF) are BNIP-3 negative. These changes, which are seen in non-necrotic cell death, or ‘para-apoptosis,’ may result from Bcl-XL resistance and/or defective calreticulin.67 Abnormal calreticulin signalling in CALRMut cases represents an alternative pathway which may disrupt megakaryocyte apoptosis through improper BNIP-3 folding and its subsequent degradation, and/or failed caspase-8 activation.28
Our data support a pathological bioprofile whereby mega-karyocyte survival in MPN is dependent upon proliferative and apoptotic mechanisms that are regulated by upstream JAK-STAT and calreticulin signalling cascades. We show that MPN with a JAK2V617F mutation had greater BNIP-3 positivity than ‘double negative’ cases. Once acquired the JAK2V617F mutation may trigger an internal apoptotic cascade as the megakaryocyte ‘recognises’ that it has accumulated irreparable DNA damage in excess.33–35 Previously attenuated BNIP-3 may then be partially upregulated to stimulate megakaryocyte apoptosis.36 ,45–48 Alternatively, the accumulation of BNIP-3 may represent a normal, remedial megakaryocyte response to counteract the anti-apoptotic effects conferred by excess Bcl-XL.36 ,45–48
Myeloproliferative megakaryocytes also showed a significant increase in SUZ12, thereby conferring either a ‘protective’ or an ‘oncogenic’ effect.56 Upregulated SUZ12 may ‘protectively’ augment the functional activation of EZH2, thereby enhancing the repression of cell cycle genes and restraining excess mega-karyopoiesis.56 ,57 ,61 Alternatively, since the upregulation of SUZ12 is not matched by a concomitant increase in EZH2, abnormal PRC2 components may form with reduced gene repression capabilities.56 Consequently, proliferation promoting genes would not be silenced and uncontrolled megakaryopoiesis would ensue through an ‘oncogenic’ mechanism. As SUZ12 megakaryocyte positivity was most marked in ET, we favour the ‘protective’ model in restraining megakaryopoiesis. With the accumulation of mutations and acceleration towards fibrosis,66 SUZ12 is depleted (as seen in MF) and the regulatory capability of the PRC2 is weakened.56 The reduction in PRC2 leads to suboptimal gene repression of proliferation and survival promoting targets,56 ,57 ,61 and enhances megakaryocyte hyperplasia in accelerating phases of MPN.
In summary, the present study demonstrates that megakaryocytic hyperplasia in MPN is driven by a combination of effects: a proliferative advantage, concurrent apoptotic failure and deterioration of regulatory epigenetic mechanisms (figure 5). In ET, where the megakaryocytes are giant with hyperlobated nuclei, they have a predominantly proliferative profile as evidenced by Ki67 expression. In contrast, myelofibrotic mega-karyocytes are dominated by impaired apoptotic functions, as indicated by increased anti-apoptotic Bcl-XL, reduced pro-apoptotic BNIP-3 and a para-apoptotic morphological appearance. Megakaryocytic expansion in MF is further enhanced by perturbed epigenetic regulatory processes affecting the PRC2. Further, dysregulated megakaryocyte apoptosis in CALRMut MPN may provide a biological explanation for the greater thrombocytosis in these patients. The above data has enhanced the pathophysiological underpinnings of megakaryocyte hyperplasia in MPN and may translate into future diagnostic benefits.
Take home messages
Megakaryocyte hyperplasia in the myeloproliferative neoplasms is due to a combination of enhanced proliferation, impaired apoptosis and dysfunctional epigenetic regulatory processes.
The megakaryocytes in essential thrombocythaemia show a trend towards a proliferative signature whereas in myelofibrosis demonstrate greater apoptotic failure.
Greater dysregulation of megakaryocyte apoptosis seen with the CALRMut genotype may be the mechanism for the higher platelet counts reported in these patients.
Acknowledgments
The authors thank Phillip Maslen and Bob Mirzai for expert technical assistance. JAJM and AA were recipients of the Cancer Council WA Honours scholarships and Leukaemia Foundation Dominic Di Giacomo Honours scholarships. JAJM also received financial support from the University of Western Australia through the Hackett Foundation Alumni Honours scholarship.
References
Footnotes
Handling editor Mary Frances McMullin
Contributors JAJM, KAF and AA performed the immunohistochemical studies. JN, GR and BG performed the molecular analyses. JAJM, KAF and KSM performed the statistical analyses. SLH and WNE performed the morphological review. RH and WNE initiated the study. All authors contributed to the writing of the manuscript.
Funding The study was supported by the Cancer Council Western Australia, Leukaemia Foundation Australia, Perpetual Foundation-Ann Helene Toakley Charitable Endowment, the Royal College of Pathologists of Australasia and the University of Western Australia.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Sir Charles Gairdner Hospital Human Research Ethics Committee (Study number 2012-094).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Raw data is available upon request.