Article Text

Abstract
Multiple myeloma (MM) is predominantly an incurable malignancy despite high-dose chemotherapy, autologous stem cell transplant and novel agents. MM is a genetically heterogeneous disease and the complexity increases as the disease progresses to a more aggressive stage. MM arises from a plasma cell, which produces and secretes non-functioning immunoglobulins. Most MM cells are sensitive to proteasome inhibitors (PIs), which have become the main drug in the treatment of newly diagnosed and relapsed MM. However, not all MM is sensitive to PIs. This review summarises the literature regarding molecular biology of MM with a focus on the unfolded protein response and explores how this could affect drug sensitivity and progression of disease.
- MYELOMA
- CANCER
- HAEMATOLOGY
- MOLECULAR BIOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Cytogenetics and mutational changes of MM
Multiple myeloma (MM) is a malignancy of the plasma cell (terminally differentiated B-Lymphocyte) characterised by proliferation of plasma cell clones.1 The disease is preceded by a pre-malignant condition monoclonal gammopathy of unknown significance (MGUS). Progression of MM to a more aggressive disease is associated with the acquisition of genetic mutations, chromosome abnormalities, copy number abnormalities and gene expression signatures. Many of these alterations are associated with changes to cell growth, apoptosis, metabolism and the epigenetics of MM cells.
Translocation of IgH gene locus (14q32 locus) occurs in about 50% of MM. It is characterised by errors of IgH switch recombination and somatic hypermutation resulting in the juxtaposition of IgH gene sequences with non-immunoglobulin DNA loci of 11q13, 4p16.3, 16q23, 6p21 and 20q11 in 60% of cases.2 ,3 Translocation of the t(4,14) locus brings oncogenes FGFR3 and MMSET under the enhancer elements of the IgH gene locus and is also associated with high expression of cyclin D2.2 T(11,14) leads to the dysregulation of cyclin D1, whereas t(6,14) leads to the overexpression of cyclin D3. The oncogenes in 16q23 and 20q11 are MAF and MAFB, respectively, and are associated with high expression of cyclin D2.4 Cytogenetic analyses of IgH translocations have served as important diagnostic and prognostic markers in MM. Translocations such as t(11;14)(q13;q32) in patients who have undergone high-dose chemotherapy and stem cell therapy are associated with improved survival outcome,5 while the t(4;14) translocation under the IgH locus has a relative poor prognosis regardless to whether patients are undergoing conventional or high-dose therapies.5
While translocations are a frequent occurrence in MM, deletions and copy number changes are just as important to the disease and in some cases just as frequent. Gains on chromosome 1 (60%) and deletion of chromasome 13 (59%) are frequently occurring characteristics among patients with MM.6 ,7 Important tumour suppressor genes such as RB1 (13q), DIS3 (13q), CDKN2C (1q) and FAF1 (1q) that are positioned within these regions are commonly affected.6 This subsequently contributes to the uncontrollable progression of the disease, by the loss of CDKN2C and RB1, leading to an unregulated cell cycle within the MM cells.8 In addition to this, the loss of FAF1 prevents MM cells to undergo apoptosis.
Single nucleotide variations, chromosomal abnormalities and epigenetic alterations are associated with the progression of MM.9–11 There is evidence that one of the driving forces behind MM progression is a result of secondary mutational changes to oncogenic pathways.11 One such pathway is the deregulation of MYC. A frequently occurring feature of MM, up to 47% of patients are seen to have an overexpression of MYC as a result of a rearrangement.11 It has also been found that the shift from MGUS to MM might be driven by MYC, indicated by its activation during this change.11 ,12 This is supported by a recent study which has found that 85% of patients with MM studied were identified as having MYC activation signalling, while being undetectable in MGUS subjects.13 However, 62.5% of patients with MGUS that progressed and developed to MM began to express MYC.13
Emerging roles of the unfolded protein response, ubiquitin proteasome system and autophagy
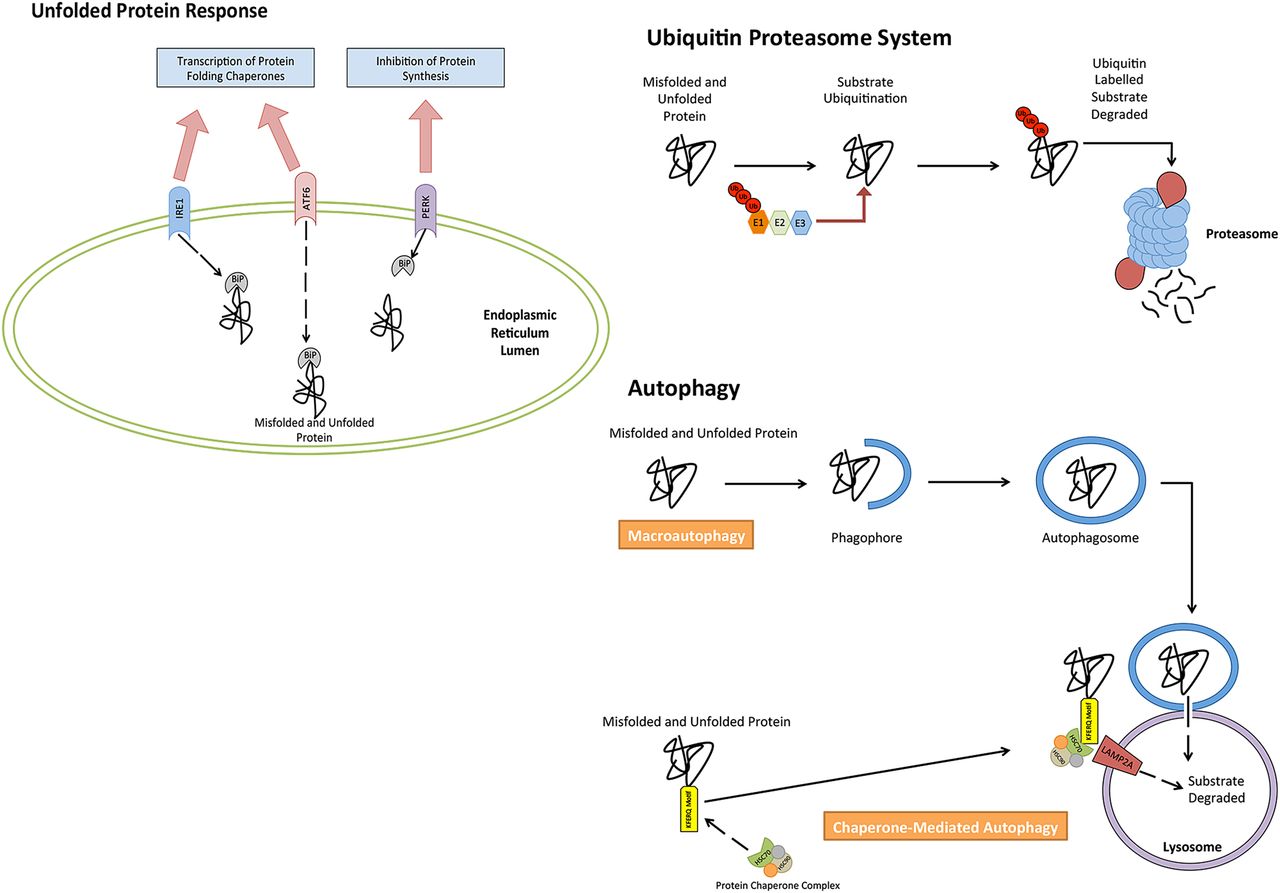
Autophagy, unfolded protein response (UPR) and ubiquitin proteasome system (UPS) are extremely versatile mechanisms responsible for maintaining cellular homeostasis. Each pathway is necessary in the management of proteostasis within cells (figure 1), and has been found to be important in MM biology. Autophagy is a cellular catabolic mechanism capable of degrading and recycling many cellular substrates, such as damaged cellular organelles and protein aggregates.14 The UPS has a similar function to autophagy; however, it strictly targets damaged or unwanted proteins that have been marked for their degradation via ubiquitin labelling of the unwanted or damaged proteins.15 ,16 Both pathways are used by the UPR,14 ,17 a pathway responsible for managing endoplasmic reticulum (ER) stress attributed to by misfolded and/or unfolded protein.18 Activation of this pathway subsequently leads to the decrease in protein synthesis, however, and an increase in the synthesis of protein folding chaperones and proteins with proteolytic functions.
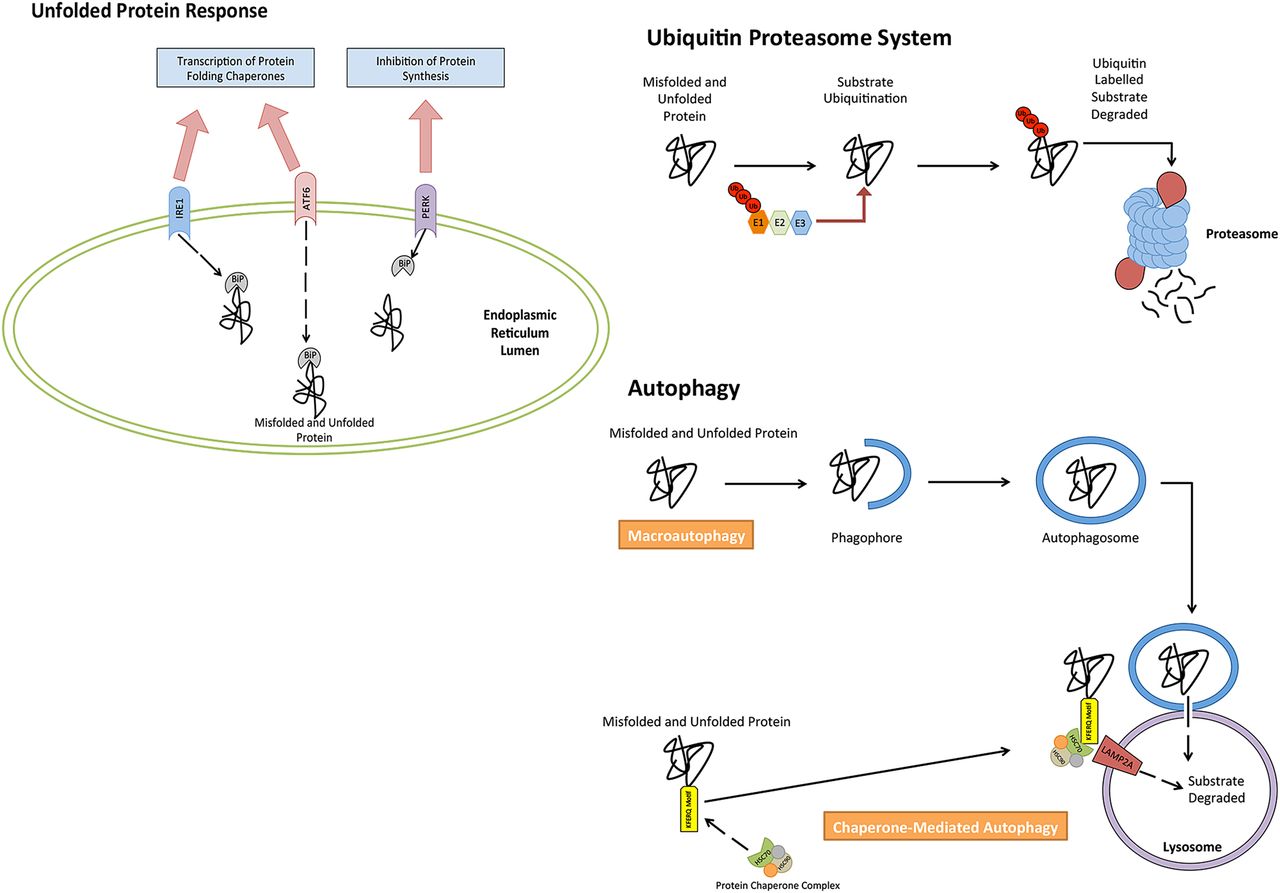
Cellular mechanisms involved in the control of proteostasis. The accumulation of unfolded and misfolded protein can be reduced through the catabolic degradation of protein through both autophagy and the ubiquitin proteasome system. Cells further rely on the unfolded protein response to alter and control protein synthesis, in order to reduce accumulating unfolded/misfolded protein.
MM cells are heavily dependent on the UPR, which is responsible for alleviating ER stress levels on the cell caused by the excessive amounts of paraprotein being produced. It is relatively more sensitive to proteasome inhibitors (PIs) compared with other cancers. This dependence on the UPR is likely to mediate a better response to the PI bortezomib. The inhibition of the 26S proteasome subsequently results in a lethal accumulation of unfolded/misfolded protein within the cell. Initially patients respond well to treatment; however, eventually the majority of patients with MM relapse and no longer respond to further treatment. Furthermore, expression levels of key regulators of the UPR, such as XBP1, appear to be significantly lower in patients as resistance increases.26 It is possible that alternative mechanisms are responsible for alleviating ER stress in resistant patients and might be a potential therapeutic target. Alternative pathways that have been studied include autophagy, deubiquitylating enzymes (DUBs) and heat shock proteins (HSPs). All three areas are likely to be important in managing and alleviating ER stress in MM, contributing to cell survival and resistance. Preliminary data are promising, with the inhibition of critical components in all three areas showing promising therapeutic potential.
The UPR is an important cellular process that occurs in response to stress placed on the ER of the cell.19 The pathways activation is a result of a number of stimuli. In MM, however, the pathway is activated by exceeded levels of unfolded and/or misfolded proteins within the ER, which surpasses the ER's ability to process.19 ,20 This molecular pathway relies on three independent stress sensors necessary for the detection of ER stress. The three stress sensors, IRE1, PERK and ATF6, are ER transmembrane proteins, which transduce ER stress signals across the ER membrane, trigging the UPR.19 ,21–23 In their inactive state, each transmembrane protein is bound the molecular chaperone binding immunoglobulin protein (BiP).24 In instances where misfolded or unfolded proteins accumulate within the ER, BiP occupying transmembrane ER proteins (inactive state) dissociate from transmembrane proteins, binding to misfolded or unfolded proteins. This, as a result, causes a conformational change to the transmembrane proteins into their active isoforms, leading to the activation of the UPR, which targets a series of downstream targets responsible for restoring cellular homeostasis and relieving ER stress.25
MM cells heavily depend on the UPR as a way to alleviate ER stress caused by the excessive production of paraprotein. For this to be achieved, the pathway relies heavily on a critical transcription factor, XBP1, a critical regulator of the UPR (downstream target of IRE1) and an important factor in plasma cell differentiation for its activation. This is achieved by the splicing of a 26 bp intron from the XBP-1 mRNA. The spliced isoform subsequently translocates to the nucleus to activate a variety of UPR target genes necessary to relieve the cell from ER stress. Low XBP1 gene expression has been found to coincide with poor responsiveness to bortezomib treatment in patients with MM, as sensitivity to bortezomib appears to increase as gene expression levels of XBP1 increases.26 Two point mutations have been identified within the gene.27 ,28 The first mutation XBP1-L167I is located within the splice site of the XBP1 gene and has been shown to prevent the splicing of XBP1 mRNA into its active spliced form in cells transfected with the mutated version, while cells which express the wild-type variant are capable of successfully splicing and activating XBP1 under ER-induced stress.27 ,28 The second XBP1 mutation XBP1s-P326R is located within the transactivation domain of the spliced XBP1 isoform and is a non-conservative missense mutation.27 Further investigation of this mutation was found to have little to no impact on the splicing of XBP1 mRNA into its active isoform.28 Reporter assays found that the transcriptional activity between the wild-type XBP1 and XBP1s-P326R-mutated variant had no significant difference under ER stress conditions.28
On further investigation, XBP1-L167I has been seen to contribute to bortezomib resistance, along with the XBP1s-P326R mutation, despite the limited impact on XBP1 splicing.27 Knockdowns of XBP1 have shown to attenuate bortezomib cytotoxicity, with spliced XBP1 found to sensitise cells to bortezomib.27 Furthermore, cells expressing either XBP1-L167I or XBP1s-P326R mutations failed to re-sensitise to bortezomib, allowing resistance to bortezomib.27
The proteasome inhibition has become the primary target for drug therapies in an attempt to treat MM. Responsible for the degradation of unfolded/misfolded proteins, its inhibition by drugs such as bortezomib subsequently results in a lethal accumulation of unfolded/misfolded protein, triggering apoptosis.29 ,30 While initially proteasome inhibition in patients with MM is effective, resistance to this drug is an often occurrence among patients with MM.30 A number of underlying contributing causes behind PI resistance in MM has been identified; however, the primary cause still remains unknown. Building evidence is starting to indicate the importance of DUBs, USP14 and UCHL5, in MM survival and possible cause behind bortezomib resistance.31 High expression levels of these two proteins have already been identified in bone marrow cells and MM cell lines of patients with MM, while having no detectable expression in normal plasma cells.31 This has indicated that both USP14 and UCHL5 could potentially be deubiquitylating misfolded/unfolded proteins in MM cells, subsequently reducing stress levels. Evidence to support such suggestions has been seen by USP14 and UCHL5 siRNA knockdowns and inhibiting the deubiquitylating activity of these enzymes by a novel 19S regulatory particle inhibitor, b-AP15. In combination, MM cells display a reduction in cell viability, along with proliferation inhibition.31 Cells that were resistant to bortezomib were also seen to overcome bortezomib resistance, becoming sensitive to the drug once more.31 These results have also been further supported by the findings of the Feng et al32 study which have shown that b-AP15 inhibits the deubiquitylating activity of USP14 and UCHL5 enzymes, triggering apoptosis in MM cell lines in a time-dependent and dose-dependent manner. Further studies by Liu et al33 further confirmed the importance of USP14 and UCHL5 in the MM malignancy, using an alternative inhibitor, Copper pyrithione (CuPT). CuPT, which also inhibits DUBs, has been shown to cause similar cytotoxic effects within MM cells as seen in other studies using b-AP15.
While most studies focus on the possible underlying causes of resistance in MM, others continue to explore the possible mechanisms responsible for managing stress levels within MM cells, despite in many instances compromising UPR activity. The primary focus has begun to explore a cellular pathway called autophagy. Autophagy is an evolutionarily conserved cellular mechanism important for maintaining cellular homeostasis.34 The degradation of such cytoplasmic components occurs within the cells’ lysosomes, the organelles of the cell that are responsible for degrading waste materials and cellular debris, facilitated by hydrolase enzymes.35 ,36
The very diverse cellular pathway of autophagy can be divided into three pathways that have distinguishable characteristics. These three pathways are macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). The primary function of macroautophagy is the elimination of damaged cell organelles or unused proteins.37 This is achieved by the formation of a double membrane around the organelle known as an autophagosome, before being degraded. Microautophagy, however, is a non-selective lysosomal degradative process, which directly engulfs cytoplasmic material by autophagic tubes through both invagination and vesicle scission into the lumen.38 CMA degrades specific cytosolic proteins through a chaperone protein complex that delivers the protein substrates to a lysosomal membrane-bound protein.39
Through a number of studies, autophagy has been identified to have a variety of pathophysiological roles in a variety of cancers including MM. A study by Kharaziha et al40 found that sorafenib-induced autophagy prevented MM cells from entering apoptosis and inhibition of the autophagic pathway resulted in cell death.40 Similar findings by Kawaguchi et al (2011) had found that inhibition of autophagy in MM enhanced the cytotoxic effect on MM cells in combination with bortezomib. Inhibition of autophagy enhances cytotoxic effects of drugs on MM cells as autophagy basal levels are relatively high in the disease as a result of elevated protein levels. Aronson et al41 has shown that induction of autophagy is prosurvival in MM cell lines and there is significant crosstalk between autophagy and the proteasomes. As autophagy is induced by inhibition of PI3K/mTOR pathway, proteasome activity is decreased. This is associated with the downregulation of the UPR genes and the PSMD14 gene, which is responsible for the binding of the ubiquinated protein and stability of the proteasome. Therefore induction of autophagy leads to proteasome inhibition indirectly. This gives reason to believe that autophagy might be a protective mechanism within myeloma cells.
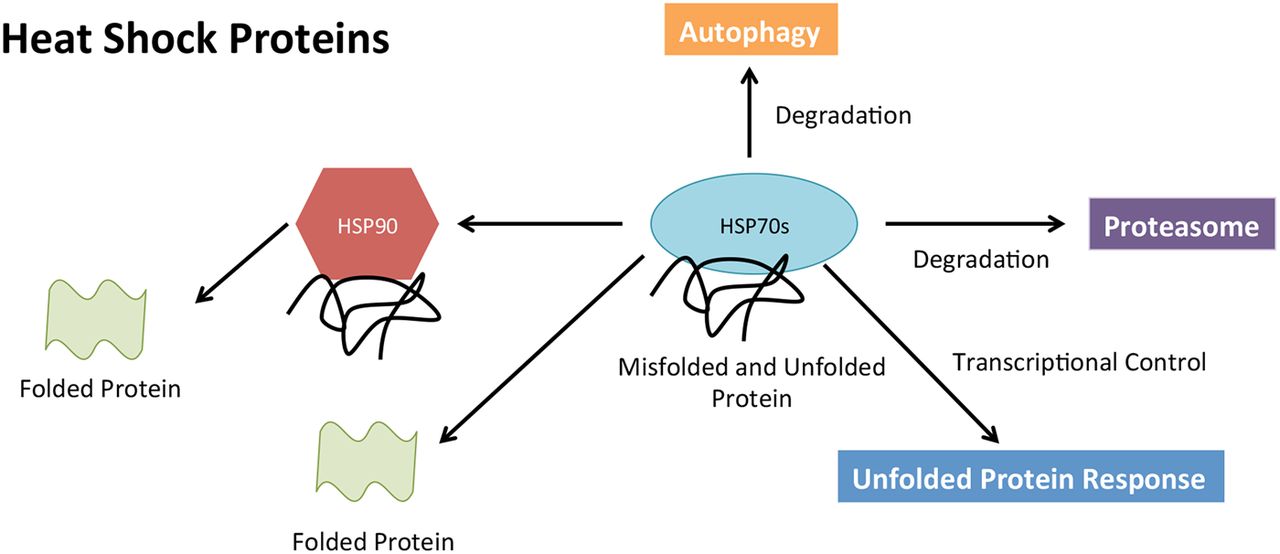
While HSPs are a focal point in many cancer types, HSP90 and heat shock conjugate 70 (HSP73) are no exception in myeloma. HSP73 (HSP70), a critical component of the CMA cytosolic chaperone complex responsible for identifying all substrates the CMA pathway degrades, is highly overexpressed in MM cells.42 Strong protein expression levels of HSP73 have been detected in the vast majority of plasma cells studied in patients with myeloma and are consistently expressed in a number of myeloma cell lines, with no detectable expression of the protein in healthy plasma cells.42 Inhibition of HSP70 in vitro leads to apoptosis of MM cells.42
Further emphasis has been placed on HSP90 as a potential therapeutic target due to its large involvement in a variety of cellular pathways (figure 2) and its cancer-promoting properties of programmed cell death inhibition and autonomous cell growth.43 Well-known client proteins of HSP90 include 60% of kinases, 30% ubiquitin ligases and 3% transcription.44 Some examples including STAT3, p53, MEK, BCR-ABL and AKT have already been established as essential contributors associated with the progression of MM and many other cancers.43 With these proteins heavily dependent on HSP90, new therapies are beginning to be developed in targeting this important protein. Such drugs include NVP-HSP99, NVP-AUY922 and Ganetespib, all of which have successfully inhibited HSP90 in vitro in myeloma and other cancers.43 ,45 ,46 Inhibition of HSP90 has been shown to be associated with increase in HSP70 expression. Davenport et al47 have shown that dual inhibition of HSP 70 and HSP90 leads to synergistic killing of MM cells in vitro. This is further supported by HSP72 and HSP73 inhibition and knockdowns in more recent studies in MM and lung cancer cell lines.42 ,48 Both HSP70 proteins support HSP90 function, contributing to MM cell survival.42
{kind=link}
{kind=link}
Heat shock proteins (HSPs) role in proteostasis. Both HSP70 and HSP90 are able to orchestrate and facilitate, with the assistance of other molecular pathways, the folding and degradation of unfolded/misfolded proteins within the cell.
Conclusion
In summary, MM is a complex disease associated with high occurrence of chromosomal, genetic and epigenetic alterations that contribute to the phenotype of the disease. The complexity of the disease is what makes understanding its biology so difficult. What is well known of the disease is that the vast majority with the disease are identified in having IgH translocations, as well as other translocations that serve as important diagnostic and prognostic markers in MM, which provide valuable insight of patients’ prognosis.
Furthermore, genetic and epigenetic alterations of tumour suppressor and oncogenes have also been identified in further altering the biology of MM, driving tumour progression. This has now led to research focusing on valuable molecular pathways that are critical to the functioning of MM. Primary focus into such pathways involved in the management of ER stress, such as the UPR, have been the focal point in drug development. Without the UPR, MM cells would subsequently succumb to a lethal accumulation of paraprotein. This has therefore led to the development of PIs, such as bortezomib. However, while initially effective, patients eventually relapse to this class of drug and become resistant.
Research has now started to stem from the UPR to other pathways and proteins, which might be contributing to drug resistance and prosurvival. Further research is needed however to establish a better understanding of what pathways and proteins are involved in contributing to drug resistance. Having said this, great interest has started to surround DUB proteins, HSPs and autophagy as contributing factors in drug resistance in MM. It is believed that they play a vital role in managing ER stress with MM cells, better and more in-depth understanding of their role in MM biology and drug resistance will lead to the development of new treatment strategies in MM.
References
Footnotes
Handling editor Mary Frances McMullin
Contributors All authors contributed equally to the review paper.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction