Article Text

Abstract
Despite its successful use in academic research, next-generation sequencing (NGS) still represents many challenges for routine clinical adoption due to its inherent complexity and specialised expertise typically required to set-up, test and operate a complete workflow.This study aims to evaluate QIAGEN's newly launched GeneReader NGS System solution in a pathology laboratory setting by assessing the system's ease of use, sequencing accuracy and data reproducibility. Our laboratory was able to implement the system and validate its performance using clinical samples in direct comparison to an approved Sanger sequencing platform and to an alternative in-house NGS technology. The QIAGEN workflow focuses on clinically actionable hotspots maximising testing efficiency. Combined with automated upstream sample processing and integrated downstream bioinformatics, it offers a realistic solution for pathology laboratories with limited prior experience in NGS technology.
- TUMOUR MARKERS
- MOLECULAR ONCOLOGY
- MOLECULAR PATHOLOGY
- LABORATORY MANAGEMENT
- METHODOLOGY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Next-generation sequencing (NGS) holds the potential to transform our understanding, treatment and management of cancer. Not only does it offer unprecedented genetic insights at a molecular level, it also enables personalised cancer treatment to maximise effectiveness. Targeted application of NGS technology allows the identification of specific mutations that affect cancer evolution and its likely response to a particular regimen, thus providing an essential tool in guiding clinical decisions.1 As such, oncologists are increasingly requesting molecular testing of cancer samples. In addition, in a clinical pathology laboratory, targeted NGS has the advantages of low sample input,1 ,2 high volume throughput and the ability to simultaneously interrogate multiple genes.3 For biopsy-based cancer diagnosis, where the tissue source is often limited, the ability to gain maximum insights from minimal sample material is particularly beneficial.2 As such, many pathology laboratories such as ours are faced with a pressing need to establish an NGS workflow to meet these requirements.
However, a number of challenges still exist against widespread adoption of NGS in clinical diagnostics laboratories. These include substantial equipment and operational costs, often complex workflows and the need for specialised bioinformatics capabilities to analyse and interpret the data for clinically meaningful use. To this end, QIAGEN recently launched the GeneReader NGS System offering a complete solution with a reduced hands-on time and fully integrated bioinformatics pipeline. This system is aimed at removing these barriers and enabling more laboratory to use the NGS technology.
To test the performance of the system, we conducted a comparison study between the GeneReader and an alternative in-house NGS platform. We tested formalin-fixed and paraffin-embedded (FFPE) clinical tumour samples taken from a range of cancer types (colorectal and gastric carcinoma, lung cancer, melanoma and gastrointestinal stromal tumours (GIST)) and processed them in two parallel workflows. Mutational status and variant allele frequencies were compared and verified via Sanger sequencing. Here we report highly consistent results from both workflows. Furthermore, we assessed the usability of the GeneReader System in a clinical pathology laboratory setting. Our experience confirms that the GeneReader NGS System is suitable for routine pathology laboratory use.
Materials and methods
Sample and DNA isolation
FFPE tumour material from various clinical sources has been collected at the Institute for Pathology at the University Hospital of Cologne. The following tumour tissue types have been considered: colorectal cancer (CRC), lung cancer, melanoma, GIST and gastric cancer (table 1). All samples were processed by the routine diagnostic pipeline of the Institute for Pathology in the years 2015 and 2016. The genomic DNA (gDNA) extracts, all characterised for the tumour/normal cells ratio and for clinical relevant mutations by the in-house NGS pipeline, based on the Illumina MiSeq, were selected as listed in table 1 for our study.
Clinical samples considered by this study with indication of the tumour percentage present in the relative formalin-fixed and paraffin-embedded material and in the targeted genes
Tumorous areas were marked by senior pathologists, and tumour nodules were scraped off from 3 to 9 µm thick sections corresponding to the H&E stained section. Automated DNA isolation from the macrodissected tumour areas has been then carried out using the Maxwell 16 FFPE Tissue low-elution volume DNA Purification Kit on a Maxwell 16 instrument (Promega GmbH, Mannheim, Germany) as described previously.4 ,5 ,6 To determine PCR accessible DNA, real-time quantitative PCR (qPCR) was performed using a standard curve of high-quality human gDNA (Takara/Clontech, Saint-Germain-en-Laye, France) ranging from 0.195 to 50 ng. qPCR targeting a 234 bp region of the HFE gene was then carried out in duplicates with 1 µL DNA, each added to a 20 µL reaction mix containing GoTaq qPCR Master Mix (Promega) and 0.4 µM of the HFE forward (TTC TCA GCT CCT GGC TCT CAT C) and reverse primer (TCG AAC CTA AAG ACG TAT TGC CC).6 ,7 The variety of different ages and origin of the FFPE samples used for this study generated different amounts of extracted DNA, as determined by qPCR (see online supplementary table S2). All 50 samples extracted passed qPCR quality control and yielded enough material for further processing. For the target enrichment step, a fixed PCR protocol with 23 or 26 cycles has been used for all samples for the MiSeq and GeneReader workflows, respectively, according to manufacturer's protocol.
supplementary data
GeneReader assay and sequencing
In total, 40 ng of each gDNA was used as template for the QIAGEN Actionable Insight Tumour Panel. The QIAGEN ATP assay amplifies 330 amplicons covering 16.7 kb, interrogating 773 unique variant positions in 12 genes of high prognostic and therapeutic relevance (KRAS, NRAS, KIT, BRAF, PDGFRA, ALK, EGFR, ERBB2, PIK3CA, ERBB3, ESR1 and RAF1) (see table 2). Targeted amplicons have been further processed to generate a library for sequencing. Libraries were prepared using the QIAGEN GeneRead DNA Library Kit and an automated protocol on a QIAcube according to the manufacturer's instructions. Both PCR-enriched DNA and GeneRead libraries were qualified and quantified using a QIAGEN QIAxcel Advanced System according to the manufacturer's instructions. The emulsion PCR and bead enrichment steps were carried out using the GeneRead Clonal Amp Q Kit and an automated protocol on a GeneRead QIAcube according to the manufacturer's instructions. Following clonal amplification, amplicon libraries were sequenced using the QIAGEN GeneRead Sequencing Q Kit and an automated protocol on a GeneReader instrument (all protocols available on http://www.qiagen.com).
Parameter and sequencing coverage results achieved in this study by the Actionable Insight Tumour Panel
GeneReader data processing
QIAGEN Clinical Insight Analyze (QCI-A) software performed the secondary analysis of FASTQ reads generated by the GeneReader consisting of alignment of reads to the hg19 reference, calling the variants, generating an interactive report for visualisation and quality control of the sequencing results as well as a summary of the data. All comparable variants have been identified by QCI-A secondary analysis pipeline for the ATP on FFPE material (ATPf), where a 5% allelic fraction cut-off is used to call variants for FFPE samples. In one case, sample Q11, in order to detect a PIK3CA variant (p.E542K) at low allele frequency (2.8%), we had to use the newly launched pipeline for liquid biopsies (ATPp), which allows a 0.5% allelic fraction cut-off.
After reviewing their data validity, variants were imported as industry standard variant call format into the QCI-interpret (QCI-I) web interface, which enables data interpretation for the previously identified variants. QCI-I then generated a per sample report for each detected variant based on the curated content of QIAGEN Knowledge Base including: summary of findings, direct link to the data source and the eventual treatment(s) recommended.
Other material and methods
Reference library construction, Illumina MiSeq sequencing and data analysis as well as Sanger sequencing are described in online supplementary material and methods.
Results and discussion
The focus of this study was to conduct a performance validation of the QIAGEN GeneReader NGS System and assess its suitability for routine use in a clinical diagnostic laboratory.
GeneReader sequencing
The laboratory set-up of the in-house established NGS workflow compared with the QIAGEN sample-to-insight NGS solution is shown in the figure 1A. Both workflows take 5 days until the diagnosis can be made. In our in-house pipeline, protocols and reagents used in the different steps of the pipeline have to be synchronised and harmoniously aligned. Therefore, our in-house solution describes a typical case scenario of a pathology laboratory where the full set-up requires prior knowledge to identify relevant kits from different vendors on one hand, but also enables high flexibility on the other hand (figure 1). The GeneReader NGS System clearly showed a close bundle of the different components in a unique product. Furthermore, the high extent of automation and integrated solutions of the GeneReader system resulted in an equal turnaround time, but in a very low hands-on time (<10 hours) (figure 1A, B).

Time course of the in-house pipeline and the GeneReader next-generation sequencing (NGS) System workflow. (A) Both workflows take a turnaround time of 5 days, but numerous automated steps reduce the hands-on time in the GeneReader NGS workflow as shown in B. #Platform and reagents were purchased from Qiagen (Hilden, Germany). 1Fluorometric quantification by the Qubit instrument and reagents (ThermoFisher Scientific, Darmstadt, Germany). (B) Cake diagram of the turnaround time using the GeneReader NGS System workflow demonstrates a low hands-on time of around 8 hours 15 min (489 min), but a hands-off time of nearly 62 hours (3714 min). QC, quality control; qPCR, quantitative PCR; QCI-A, QIAGEN Clinical Insight Analyze; QCI-I, QCI-interpret.
GeneReader sequencing data analysis
The ATP assay was developed to focus only on clinically relevant mutations as included in approved therapeutics labels, major professional practice guidelines and active late-stage clinical trials. Thus, this approach results in a selection of genes and variants with an unparalleled level of direct clinical relevance. The integrated bioinformatics pipeline QCI-A allows a streamlined analysis of the sequences and identification of genetic aberrations, while providing intrinsic quality control measures to ensure confidence in calling of the variants. Of note, 16 of the 50 samples tested did not meet the industry standard 500× coverage threshold. Yet the GeneReader still successfully mapped the reads and correctly identified the variants, as summarised in table 2. In total, there were 56 mutations with clinical relevance positively identified (see online supplementary table S2), which showed an average and median hotspot coverage of around 5000× and 7650×, respectively (table 2).
Confirmation of variants by MiSeq and Sanger sequencing
This study considered 50 clinical DNA samples processed by the routine diagnostics workflow of the laboratory for molecular pathology (Institute of Pathology at the University Hospital of Cologne). Hereby, we included samples from different origin (table 1), such as CRC, lung cancer, melanoma, GIST and gastric cancer. The samples also have been selected for their capacity to represent well-characterised variants with a clinical impact and to represent tumours with different tumour to normal cell ratios as shown in online supplementary table S2.
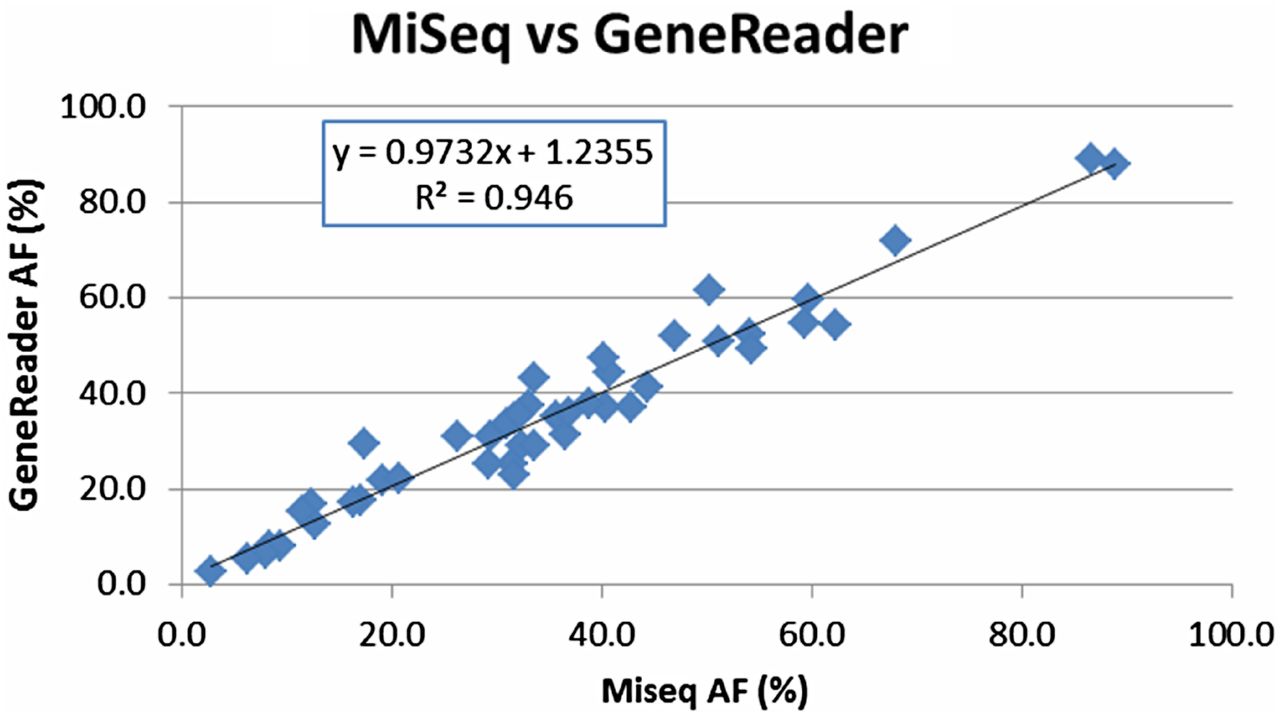
To ensure maximal sample consistency, the same DNA extractions were used for MiSeq or Sanger confirmation. Importantly, we observed a 100% agreement between GeneReader NGS system and the previously used MiSeq pipeline or the Sanger sequencing (see online supplementary table S2). For nine GIST samples with PDGFRA and KIT mutations (Q39–46), only Sanger sequencing confirmation data were available (see online supplementary table S2) with no direct variant allele frequency information. For the remaining 45 samples, allele frequency mapping showed an extremely high degree of linear correlation (R2=0.946) between the GeneReader System and the MiSeq pipeline (figure 2). Thus, importantly the concordance between the platforms is observed in a wide range of allele frequencies, from <5% to >90% (cf. figure 2).

{kind=link}
{kind=link}
Concordance between the in-house workflow using the MiSeq platform and the semiautomated GeneReader System. Direct allele frequency (AF) comparison between GeneReader and MiSeq. A high concordance (R2 =0.946) was achieved for 45 clinical variants.
Furthermore, we used the QCI-I software to assign clinical relevance and functional impact of these variants. This is a bioinformatics database built and continuously updated by a content curation team. This tool enables any laboratory to consider potential courses of action based on specific variants identified, by providing real-time information on approved or guideline-recommended therapeutics and active clinical trials. Such correct interpretation of tumour molecular changes is very important,8 in particular, because knowledge would otherwise be impractical for an individual laboratory to accumulate and update. For example, in addition to the well-known mutants in BRAF and KRAS, the activating p.Y772_A775 duplication in exon 20 of the ERBB2 (Her2) gene was recognised in three samples. For this ERBB2 (Her2) alteration, it was recently shown that patients with lung cancer, harbouring this mutation, benefit from treatment with ERBB2 inhibitors, such as afatinib, neratinib, pelitinib and dacomitinib.9 ,10 ,11
Take home messages
Targeted NGS by the semiautomatic workflow of the GeneReader System
Proved a low hands-on time and
Showed a high run performance and a high accuracy in mutation calling.
The integration of an automatic data transfer, analysis and interpretation,
Ending by the report of the mutant with the potential pathogenic impact,
Makes it as an easily accessible technology for molecular pathology laboratories.
Supplementary materials
Abstract in German
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Abstract in German - Online abstract
Footnotes
UK and CH contributed equally.
Handling editor Runjan Chetty
Contributors UK and HA performed the GeneReader workflow, whereas CH, SM-B performed NGS by the conventional in-house workflow. CH, II and UK validated data by Sanger sequencing, and AF, RB and MO performed data interpretation and illustration. RHB, AF, YK, CH and MO wrote the manuscript, which was then reviewed by all authors.
Competing interests The University Hospital of Cologne, Institute of Pathology is in a collaboration agreement with Qiagen (Hilden, Germany). Reinhard H Büttner is on the Advisory Board of Qiagen The authors declare no other conflict of interest. As of December 2016, the sequencing chemistry used in this study is not available in the USA.
Provenance and peer review Not commissioned; externally peer reviewed.