Article Text

Abstract
RNF43 (E3 ubiquitin-protein ligase RNF43 or RING-type E3 ubiquitin transferase RNF43) functions as a tumor suppressor, by exerting a predominant negative feedback mechanism in the Wnt/β-catenin signaling pathway. RNF43 inhibits Wnt/beta-catenin signaling by ubiquitinating Frizzled receptor and targeting it to the lysosomal pathway for degradation. Loss of function of RNF43 results in decrease/lack of degradation of Frizzled with enhancement of Wnt/β-catenin signaling pathway. Mutations of RNF43 have been reported in different cancers. We describe the structure of RNF43, its function and most frequent mutations in different cancers.
- gi neoplasms pancreatic
- cancer
- colorectal cancer
Statistics from Altmetric.com
Introduction
RNF43 (E3 ubiquitin-protein ligase RNF43 or RING-type E3 ubiquitin transferase RNF43) and related ZNRF3 belong to a small set of proteins, members of Goliath and Godzilla families, which bear the characteristic sequence of an ubiquitin ligase and of a transmembrane region.1
RNF43 and ZNRF3 comprise a moderate sequence conservation of 39% identity between the two proteins.2
RNF43 functions as a tumour suppressor that inhibits Wnt/β-catenin signalling by ubiquitinating Frizzled receptor and targeting it to the lysosomal pathway for degradation.
Structure
The RNF43 gene is highly conserved in vertebrates (chimpanzee, Rhesus monkey, dog, cow, mouse, rat and chicken); it is mapped to the long arm of chromosome 17 at position 23.2 (17q23.2). The peptide sequence of the gene is encoded by 11 exons spanning 60 kb.3
A regulatory region containing two Wnt-responsive elements (WREs) has been identified in intron2 (figure 1). The expression of RNF43 is regulated by the canonical Wnt/β-catenin pathway through binding of the WREs with T cell factor 4 (TCF4)/β-catenin complex.4

Diagram of the RNF43 gene. WRE1 and WRE2 are two Wnt-responsive elements (WREs) identified in intron2.
The protein has two isoforms. Isoform 1 has two variants: variant 1 represents the longest transcript and encodes the longer isoform; variant 2 uses an alternate splice site in its 5′ UTR compared with variant 1. Both variants 1 and 2 encode the same protein.5 Isoform 2, variant 3, has multiple differences, compared with variant 1. These differences result in a distinct 5′ UTR and cause translation initiation at a downstream start codon, compared with variant 1. The encoded protein has a shorter N-terminus, compared with isoform 1.5
The protein consists of 783 amino acids and has a molecular mass of 90 kD.6
RNF43 resembles a type I transmembrane receptor with an N-terminal region with a signal peptide and an extracellular domain, followed by a single membrane spanning region and a C-terminal region, which includes the intracellular RING-type E3 ligase domain (figure 2).

Diagram of RNF43 protein. RNF43 protein includes an extracellular domain with signal peptide (SP) and protease-associated (PA) domain, a transmembrane domain and an intracellular RING-type E3 ligase domain (R). Two hot spots mutations are indicated on the right.
The extracellular domain comprises a single globular domain of 150 amino acids. This domain is a variant of the protease-associated (PA) domain fold, and it has been identified in various proteases families and in a variety of receptors involved in protein trafficking both in plants and animals. Several studies have shown that the PA fold is a putative protein recognition domain in a distinctive set of transmembrane ubiquitin ligases, which function in the endocytic pathway and at the cell surface.1
The extracellular domain appears to be the prime candidate for a role in the targeting of the Wnt receptor Frizzled for ubiquitination by interaction with the N-terminal cysteine-rich domain of Frizzled with Dishevelled as adaptor protein.7
This extracellular domain is also involved in the removal of RNF43 from the cell surface and consequent increase in Wnt activity.1
The cytoplasmic RING domain of E3 ubiquitin transferase functions as a protein ubiquitylation and serves as a mediator bringing the ubiquitin-charged E2 ubiquitin-conjugating enzyme and the acceptor protein together to enable the direct transfer of ubiquitin.8
Localisation
Endogenous RNF43 is found in the nucleus of human intestinal crypt and colon cancer cells.9 It has been shown to reside in the inner nuclear membrane, the nuclear periphery and nucleoplasm,10 the endoplasmic reticulum6 and on the surface cell membrane where it interacts with Frizzled.8 The presence of the signal peptide determines the distribution of RNF43 to the plasma membrane, while its lack results predominantly in the cytoplasmic dislocation.8
RNA-seq performed on human tissue samples showed RNF43 gene expression in several normal non-neoplastic organs, with high levels in duodenum, small bowel, colon and prostate samples11 (figure 3).

RNF43 tissue-specificity expression by RNA-seq performed on tissue samples from 95 human individuals representing 27 different tissues. RPKM, reads per kilobase of transcript per million mapped reads).
Function
RNF43 is associated with the Wnt receptor complex and inhibits Wnt signalling by promoting the turnover of Frizzled. Wnt proteins are secreted glycoprotein ligands, which control cell proliferation, migration, cell fate specification and polarity formation. The canonical Wnt signalling pathway regulates gene expression modulating the transcription cofactor β-catenin. Wnt proteins can also regulate the β-catenin-independent planar cell polarity pathway. Frizzled proteins serve as a core receptor of Wnt proteins in both these pathways. The Wnt proteins interact with different coreceptors to activate downstream signalling pathways. They bind to coreceptor low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6) in the Wnt/ β-catenin pathway. Several mechanisms are involved in the regulation of the Wnt pathway and RNF43 adds furthermore control and is itself subject to modulation.1
RNF43 plays a role in mediating the ubiquitination, endocytosis and subsequent degradation of Wnt receptor complex components Frizzled. It interacts with the Frizzled family forming a specific complex with Frizzled and LRP5/6 receptors, mediating ubiquitination of specific lysine residues in the cytoplasmic loops of these receptors. The ubiquitination results in the removal of Wnt receptors from the cell surface with consequent downregulation of Wnt signalling (figure 4).

Diagram representing the interaction between RNF43 and Frizzled, which results in ubiquitination, endocytosis and subsequent degradation of Wnt receptor complex components Frizzled. The Wnt proteins interact with the Frizzled and LRP5/6 to activate Wnt/β-catenin signalling. β-catenin enters the nucleus where it interacts with TCF4 and induces the expression of RNF43 (A). RNF43 migrates to the surface membrane where it interacts with Frizzled and Dishevelled (B) and induces ubiquitination and degradation of Frizzled (C). LRP5/6, low-density lipoprotein receptor-related protein 5 or 6; TCF4, T cell factor 4.
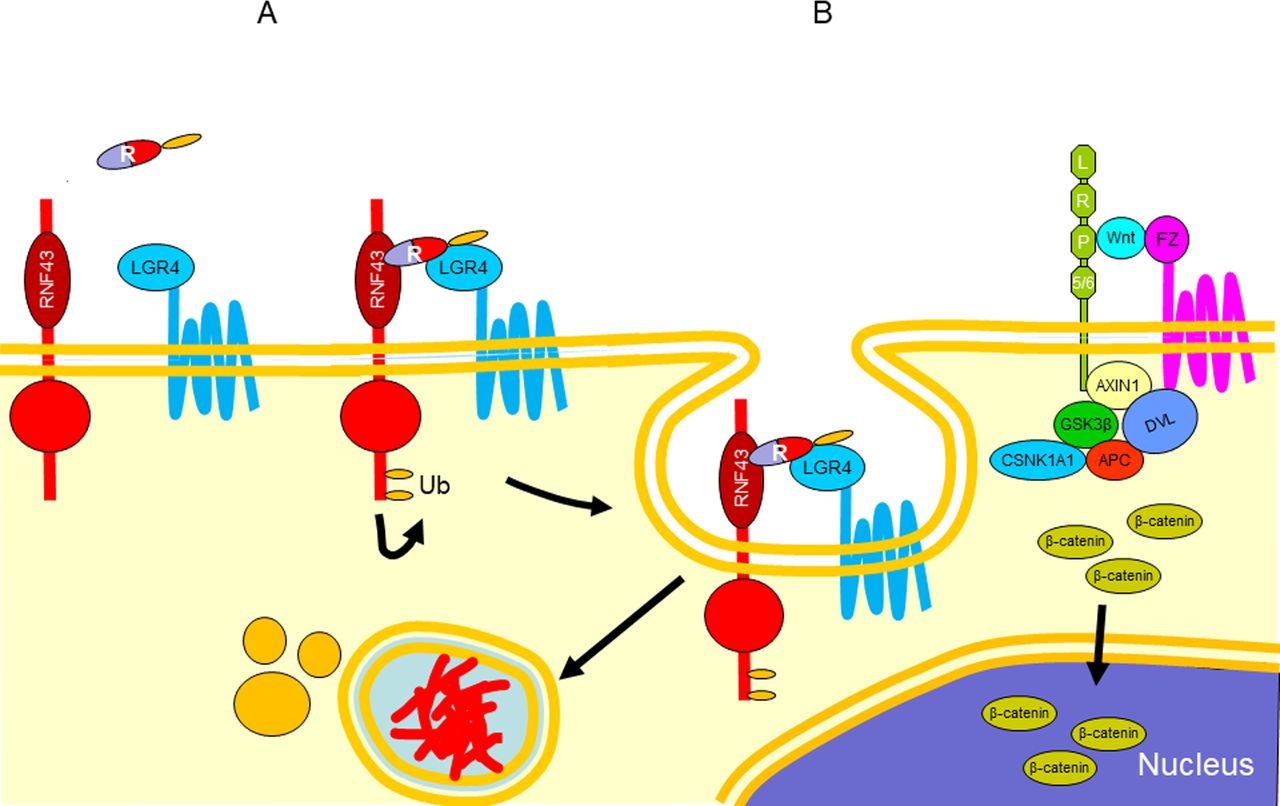
RNF43 can itself be targeted for removal from the cell surface by interacting with the ligand Respondin, a secreted protein agonist of the canonical Wnt signalling, and forming a tertiary complex with leucine-rich repeat containing G protein-coupled receptor 4/5 (LGR4/5), which induces autoubiquitination and membrane clearance of RNF43, resulting in increased cell surface level of Frizzled. The removal of RNF43 results in reaccumulation of Wnt at the cell surface with consequent enhancement of Wnt signalling (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Diagram representing the removal of RNF43 from the cell surface by interacting with the ligand Respondin (R) and LGR4/5, which induces ubiquitination (A) and degradation of RNF43 (B). LGR4/5, leucine-rich repeat containing G protein-coupled receptor 4/5.
The PA domain of RNF43 would be involved in the interaction with Respondin.
It has been shown that RNF43 physically interacts with TCF4 in cells and tethers TCF4 to the nuclear membrane, thus silencing TCF4 transcriptional activity even in the presence of constitutively active mutants of β-catenin.9
RNF43 interacts with NEDD-4-like ubiquitin-protein ligase-1 (NEDL1) and regulates p53-mediated transcription. RNF43 also interacts with p53 and suppresses transcriptional activity of p53.12 It may also be involved in cell growth control potentially through the interaction with HAP95,6 a chromatin-associated protein interfacing the nuclear envelope.
In the liver, metabolic zonation requires a Wnt/β-catenin signalling gradient, and the Respondin-LGR4/5-RNF43 module functions as a master regulator of Wnt/β-catenin-mediated metabolic liver zonation and is a hepatic growth/size rheostat during development, homeostasis and regeneration.13
Function of RNF43 in cancer
RNF43 affects a prevailing negative feedback mechanism in the Wnt/β-catenin signalling pathway. Activation of Wnt pathway results in the expression of RNF43, which turns into the degradation of Wnt receptors on the cell surface, resulting in the inactivation of Wnt/β-catenin signalling pathway. In cancer cells, a possible way to have a permanent activation of the Wnt/β-catenin signalling pathway is by inactivation of RNF43 through mutations. Loss of function of RNF43 results in decrease/lack of degradation of Frizzled with enhancement of Wnt/β-catenin signalling pathway. Another possible mechanism of the RNF43-mediated inhibition has been proposed. RNF43 is located on the nuclear membrane and tethers TCF4 to the nuclear membrane, thus silencing TCF4 transcriptional activity.9 Mutations of RNF43 would release TCF4 allowing acting as a transcription factor.
RNF43 in serrated colorectal lesions
There have been several recent studies exploring the role of RNF43 in sessile serrated adenomas (SSA) and traditional serrated adenomas (TSA).
Yan et al 13 explored serrated polyposis families and showed that 25% of families with this syndrome harboured germline mutations in RNF43. Somatic mutations, on the other hand, were noted in 34% of sporadic SSA and TSA. None of the hyperplastic polyps examined in this study displayed RNF43 mutations. Furthermore, there was a strong association with RNF43 mutations in MLH-1 methylated microsatellite unstable colorectal cancers, with 85% of such cases showing RNF43 mutations. In contrast, this dropped to only 33% in the MLH-1 non-methylated cancer cases.13
The study by Tsai et al 14 examined RNF43 mutations in both SSA and TSA. In this study only 2 of 20 (10%) of SSA and 10 of 36 TSA without dysplasia (27.8%) harboured RNF43 mutations. In addition, 7/36 (19.4%) of TSA with ‘cytological dysplasia’ also showed RNF43 mutations. They also examined a cohort of BRAF mutated, microsatellite stable colorectal cancers and found 9 of 31 cases (29%) to be RNF43 mutated.14 By way of comparison, none of the tubular or tubulovillous adenomas examined showed any RNF43 mutations. This study concluded that RNF43 and BRAF mutations were closely associated, while RNF43 was inversely related to KRAS mutations in TSA with and without dysplasia.14 They further concluded that RNF43 mutation is an early and specific molecular event in the serrated pathway, in particular in TSA and BRAF-mutated microsatellite stable colorectal cancers.14
Using targeted next-generation sequencing, Hashimoto and colleagues15 examined SSA with (46 cases) and without dysplasia (45 cases). A striking disparity was found: in SSA with dysplasia RNF43 truncating mutations were found in 58% of cases, while only 7% of SSA without dysplasia contained RNF43 mutations.15 These changes were linked to the Wnt signalling pathway as the SSA with dysplasia also showed nuclear expression of β-catenin and overexpression of myc proteins.15 Additionally, MLH-1 protein was lost in 14/46 (30%) of SSA with dysplasia, and in these 14 cases, 86% had frameshift mutations of RNF43.15 The authors concluded that Wnt pathway genes, in particular RNF43, are responsible for dysplasia in SSA and are associated with loss of MLH-1 as a result of MLH-1 promoter methylation (table 1).
Mutations in RNF43
Sekine et al 16 used targeted next-generation sequencing and reverse transcriptase in a series of TSA and found that 24% showed RNF43 mutations.
In summary, RNF43 mutations are important in the serrated pathway, with SSA and TSA harbouring inactivating mutations. More specifically, RNF43 mutations are associated with the advent of dysplasia in these serrated polyps. RNF43 mutations are not present in hyperplastic polyps, sporadic tubular or tubulovillous adenomas. In colorectal carcinoma, RNF43 is associated with BRAF mutations and more prevalent in microsatellite unstable cases with loss of MLH-1 protein expression from MLH-1 promoter methylation.
RNF43 is exclusively and frequently altered in Lynch syndrome-associated adenomas (52%). In addition, Lynch syndrome-associated adenomas show variable mutation profiles depending on the mismatch repear (MMR) status. RNF43 mutations are exclusive to MMR-deficient adenomas (66%) and are mostly frameshift mutations affecting mononucleotide repeats. These MMR-deficient adenomas harbour frequently APC frameshift mutation. On the other hand, MMR-proficient adenomas show more frequently APC or CTNNB1 mutations, which are indistinguishable from those present in sporadic adenomas.17 Lynch syndrome-associated adenocarcinomas frequently have RNF43 mutation profile similar to adenomas.17 Frameshift mutations are consistent with the consequence of MMR deficiency. These RNF43 mutation patterns have also been reported in sporadic MMR-deficient colorectal adenocarcinomas18 (table 1).
RNF43 mutations are found in 9 of 24 (37.5%) Lynch syndrome colorectal adenocarcinomas. The majority of mutations were frameshift deletions (p.Gly659fs) (29%); two cancers (2/24, 8%) from one patient harboured frameshift mutations at codon R117 (p.Arg117fs) within exon 3.19
RNF43 is a frequently mutated gene in sporadic colorectal adenocarcinomas (18.9%). The mutations identified are small insertions or deletions (p.Gly659fs and p.Arg117fs) typical of MSI tumours. Truncating mutations of RNF43 are statistically linked to the microsatellite high (MSI-H) phenotype. In this group of colorectal adenocarcinomas, RNF43 is mutated in 79.7%.18 This association suggests that the MMR deficiency provides a favourable environment for major RNF43 frameshift mutations and confer a fitness advantage to the cancer cells in which they occurred18 (table 1).
Additional data show that RNF43 is mutated in 47/54 (87%) of BRAF mutant/MSI and 8/33 (24%) BRAF mutant/microsatellite stable adenocarcinomas compared with only 3/79 (4%) BRAF wild-type adenocarcinomas (p<0.0001). The RNF43 frameshift mutation, p.Gly659fs, occurred in 80% BRAF mutant/MSI cancers.20
RFN43 in gastric adenocarcinomas
RNF43 mutations have been reported in 3 of 37 (8.1%) gastric adenocarcinomas21; however, they are more frequent id MSI in comparison to microsatellite stable (MSS) (54.6% vs 4.8%22 and 33% vs 3%23) (table 1).
RNF43 in pancreatic cystic lesions and adenocarcinoma
A series of cystic lesions of the pancreas were examined for RNF43 mutations.24 Inactivating mutations, including loss of heterozygosity, were found in six of seven (85.7%) intraductal papillary mucinous neoplasms (IPMN). RNF43 mutations were identified in 21% and 18% of non-invasive and invasive IPMNs, respectively.25 In pancreatic mucinous cystic neoplasms (MCN), four of eight cases (50%) displayed intragenic mutations (three cases), and one case showed loss of heterozygosity. None of the serous cystadenomas or solid pseudopapillary neoplasms examined in this series showed RNF43 mutations.24
Jiang et al 26 concluded that mutational inactivation of RNF43 in pancreatic adenocarcinoma cell lines confers Wnt dependency in these cancers, and hence Wnt pathway members, including RNF43, could serve as biomarkers for Wnt pathway inhibition.
RNF43 in ovarian mucinous tumours
In a similar fashion to MCNs of the pancreas, Ryland et al 27 showed that 2 of 22 (9%) of borderline mucinous ovarian tumours and 6 of 29 (20.7%) of mucinous cystadenocarcinomas contained RNF43 mutations.
RNF43 in endometrial adenocarcinomas
RNF43 is frequently mutated in endometrial adenocarcinomas in 18.1%. Truncating mutations are more frequent, with the p.Gly659fs variant accounting for 47.3% and the p.Arg117fs variant for 3.6% of the alterations. The frequency of RNF43 mutations is higher in MSI-H adenocarcinomas (50.7%).18
Other cancers
RNF43 mutations are found in 9.3% of the liver fluke-associated cholangiocarcinoma.28
RNF43 mutations have not been reported in melanoma,29 breast cancer,30 or head and neck malignancies.31
Take home messages
RNF43 exercises a powerful negative feedback on the Wnt/β-catenin signalling pathway.
Mutations of RNF43 result in enhancing this signalling pathway.
Hot spot mutations, mainly frameshift (p.Arg117fs and p.Gly659fs), are found in MMR-deficient colorectal SSA, adenomas and adenocarcinomas.
References
Footnotes
SS and RC contributed equally.
Handling editor Cheok Soon Lee.
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.