Article Text

Abstract
SIFD describes a heritable, syndromic condition characterised principally by sideroblastic anaemia (SA) with immunodeficiency, fevers and developmental delay, arising in mutations within the TRNT1 gene. Other clinical manifestations of SIFD include cardiomyopathy, seizures, sensorineural hearing loss, renal dysfunction, metabolic abnormalities, hepatosplenomegaly and retinitis pigmentosa.
Presentation of SIFD is variable but typically in early childhood with SA or with fever. In this report, we extend the described SIFD phenotype. We describe a kindred in which the index case presented with fetal hydrops, and early neonatal death, and the second child had severe anaemia at delivery. Both cases had prominent extramedullary erythropoiesis and numerous circulating nucleated red blood cells.
- congenenital anaemia
- sideroblastic anaemia
- fetal hydrops
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
SIFD describes a syndromic condition characterised principally by sideroblastic anaemia (SA) with immunodeficiency, fevers and developmental delay. Other clinical manifestations of SIFD include cardiomyopathy, seizures, sensorineural hearing loss, renal dysfunction, metabolic abnormalities, hepatosplenomegaly and retinitis pigmentosa.1 2 SIFD has recently been found to be due to mutations in TRNT1. This is a highly conserved gene through evolution and knockout of the gene in animal models is lethal. TRNT1 catalyses the addition of a terminal CCA trinucleotide residue to nuclear and mitochondrial tRNA and this residue binds the amino acid to the tRNA and delivers it to the ribosome during protein biosynthesis. TRNT1 is therefore essential for maturation and function of all cellular tRNAs.
The presentation is variable but is typically in early childhood with SA or with fever. We describe two siblings where the presentation was with severe anaemia, marked leucocytosis and hugely elevated numbers of circulating nucleated red cells. This is more typically the clinical presentation of a haemolytic anaemia. The index case died very early and postmortem (PM) examination revealed massive extramedullary haemopoiesis with iron overload. The subsequent male child had a similar haematological presentation but additionally had ambiguous genitalia among other congenital anomalies.
Case report
Case 1
Following a routine pregnancy with normal routine antenatal scans (12 and 20 weeks), a female infant was born at 38 weeks’ gestation, with a weight of 2.890 kg. The parents were non-consanguineous, and there was no significant family medical history. The patient was born in poor condition, with poor response to resuscitation requiring endotracheal intubation at 3 min of age and admission to neonatal intensive care.
The patient was profoundly anaemic, with jaundice and massive hepatosplenomegaly. The white cell count was raised with numerous circulating nucleated red blood cells (NRBC). Despite appropriate blood and fluid resuscitation, she deteriorated, with progressive jaundice, multiorgan failure and evidence of intracranial bleeding. With consensus from the family, care was withdrawn and the patient died at around 40 hours of age.
PM investigations
PM investigation confirmed a female infant weighing 3.48 kg (50–75th centiles, 39+1 weeks corrected) with a head circumference of 33 cm (25th centile, 39+1 weeks corrected).
No congenital abnormalities or dysmorphic features were identified. Generalised subcutaneous and soft tissue oedema was noted, with a generalised purpuric skin rash and jaundice. Internal examination revealed peritoneal, pericardial and pleural effusions. Haemorrhagic petechiae were seen on the anterior and posterior aspects of the heart and subpleural areas, adrenals, liver and spleen capsule. No structural cardiac anomalies were noted. Significant hepatic and splenic enlargement, and marked splenic congestion were identified, without concurrent structural abnormalities. All other intra-abdominal organs were normal in terms of structure and size.
Examination of cranial cavity and brain revealed bilateral ventricular enlargement/dilatation secondary to intraventricular haemorrhage (IVH) was present, with white matter necrosis and haemorrhagic foci within the basal ganglia.
Histology revealed widespread extramedullary haematopoiesis, with accumulation of myeloid precursor cells and NRBCs within the adrenal glands, thymus, spleen, diaphragm, microvasculature, kidneys, epicardium, small and large intestinal mucosa, liver, lungs and skin (figures 1 and 2). Bone marrow histology revealed an excess of erythroid and myeloid precursor cells, with focal hemosiderin but no definitive sideroblasts evident on Perl’s stain.
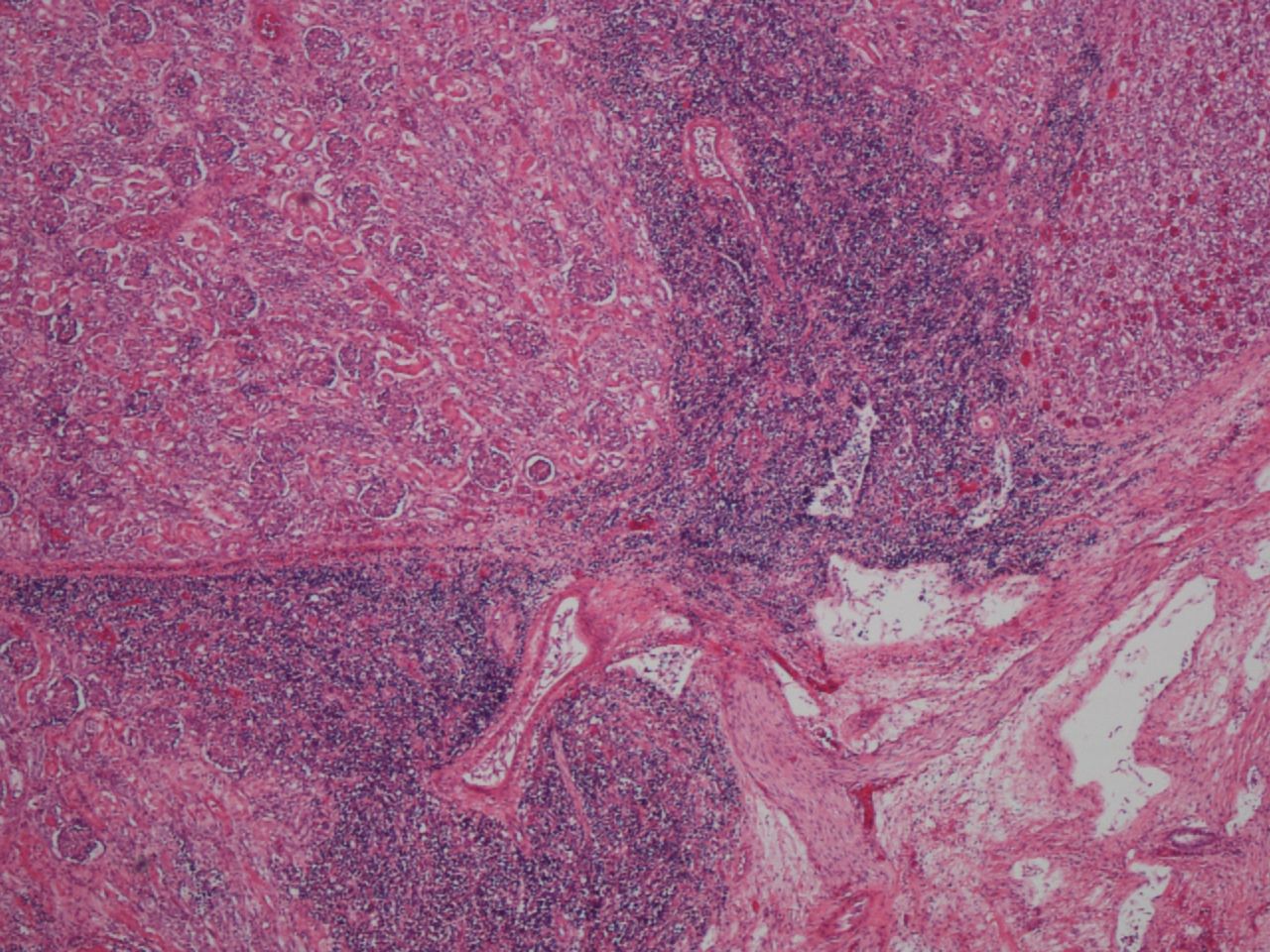
Extramedullary haemopoiesis, with nodular involvement of renal parenchyma (H&E stain, original magnification ×4).
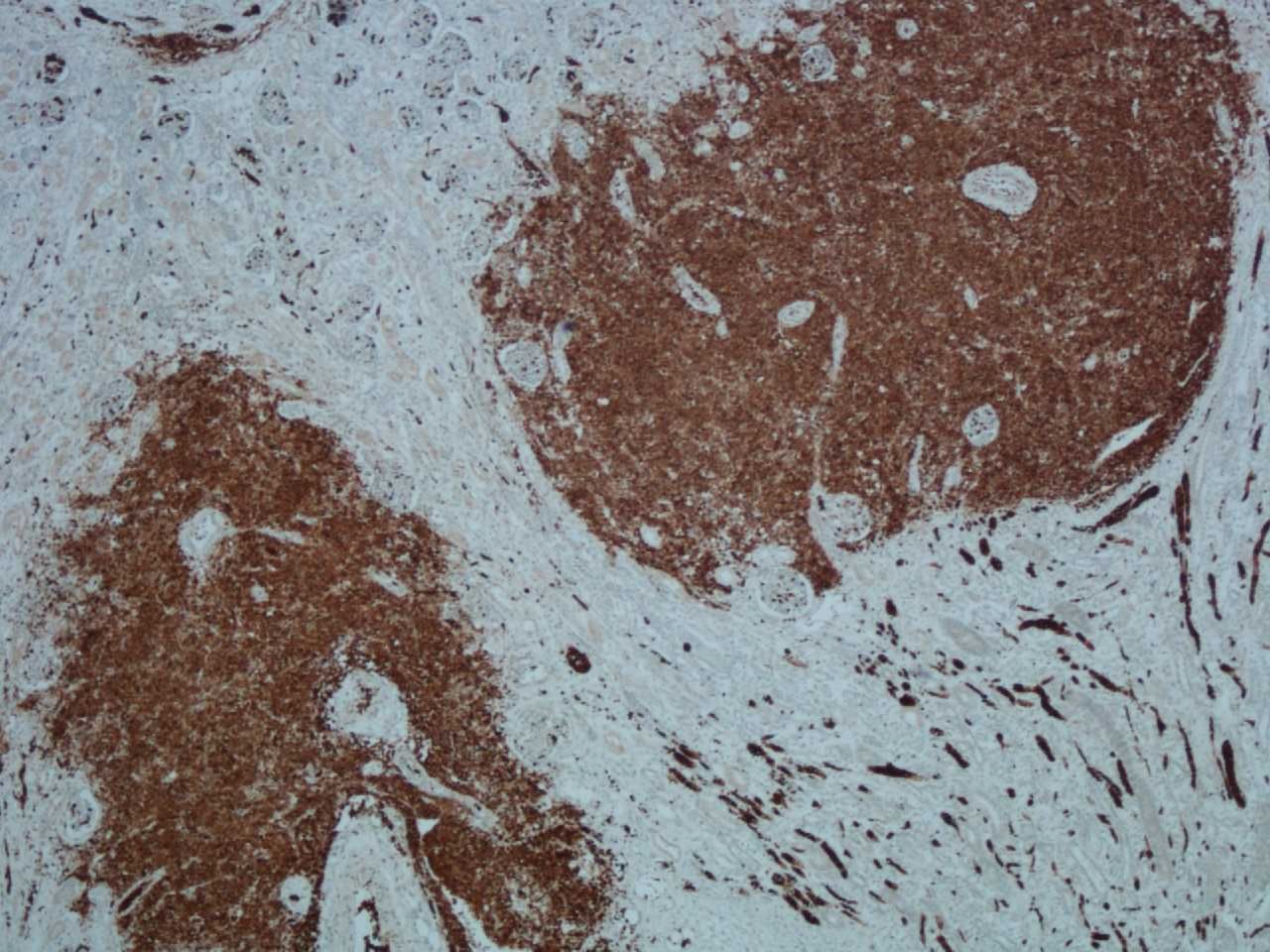
Immunohistochemistry for glycophorin highlights blood cells (red blood cells) and erythroid precursor cells within the renal parenchyma (original magnification ×4).
Macroscopic and histological placental examination demonstrated features compatible with placental hydrops. The placenta was large, pale and friable weighing 1265 g (normal placental weight 50th centile at 38 weeks of gestation is 499 g). Histology revealed marked villous hydrops and generalised villous immaturity, and diffuse extramedullary haemopoiesis with large clumps of erythropoietic cells (NRBC) and myeloid precursors within, plugging the fetal vessels, the chorionic plate vessels and the umbilical cord vessels (figures 3 and 4).

Abundant haematopoietic cells in the fetal capillaries. Fetal nucleated red blood cells are particularly prominent (H&E stain, original magnification ×40).

Placental hydrops demonstrating immature oedematous and enlarged villi with many nucleated red blood cells (H&E stain, original magnification ×4).
Summary
Overall, the clinical picture represented generalised hydrops with widespread cutaneous purpura, marked hepatomegaly and abdominal distension, and widespread extramedullary haematopoiesis. Intracranial lesions represented chronic changes secondary to intrauterine anaemia, and acute changes (eg, IVH/ventricular dilatation) secondary to profound hypoxia and disseminated intravascular coagulopathy.
The cause of death was recorded as fetal hydrops secondary to congenital anaemia of unknown cause. Neither perinatal nor PM investigation identified an underlying cause for the congenital anaemia.
Case 2
A male infant was born at 36+6 weeks’ gestation via normal vaginal delivery, following induction of labour for poor cardiotocograph traces. The pregnancy had been intensely monitored on ultrasound from 20 weeks, when short long bones, a possible cardiomyopathy and dilated bowel loops were seen. Cardiac scanning normalised, with bone growth progressing along centiles. A persistent but stable fetal tachycardia was noted at 30 weeks. There was no evidence of hydrops antenatally or at delivery.
The birth weight was 2.632 kg. Neonatal blood counts demonstrated anaemia with elevated white cell count, and prominently circulating NRBCs, suggesting extramedullary haemopoiesis. There was mild proximal upper limb shortening and penoscrotal hypospadias and microphallus, but bilaterally palpable testicles, with subsequent normal male 46 XY karyotype. The neonatal period was characterised by congenital anaemia, conjugated hyperbilirubinaemia, meconium ileus requiring ileostomy, gastric perforation requiring surgical repair, and cardiac anomalies including right ventricular hypertrophy with asymmetric septal hypertrophy and patent ductus arteriosus. Adrenal calcification was noted on X-ray.
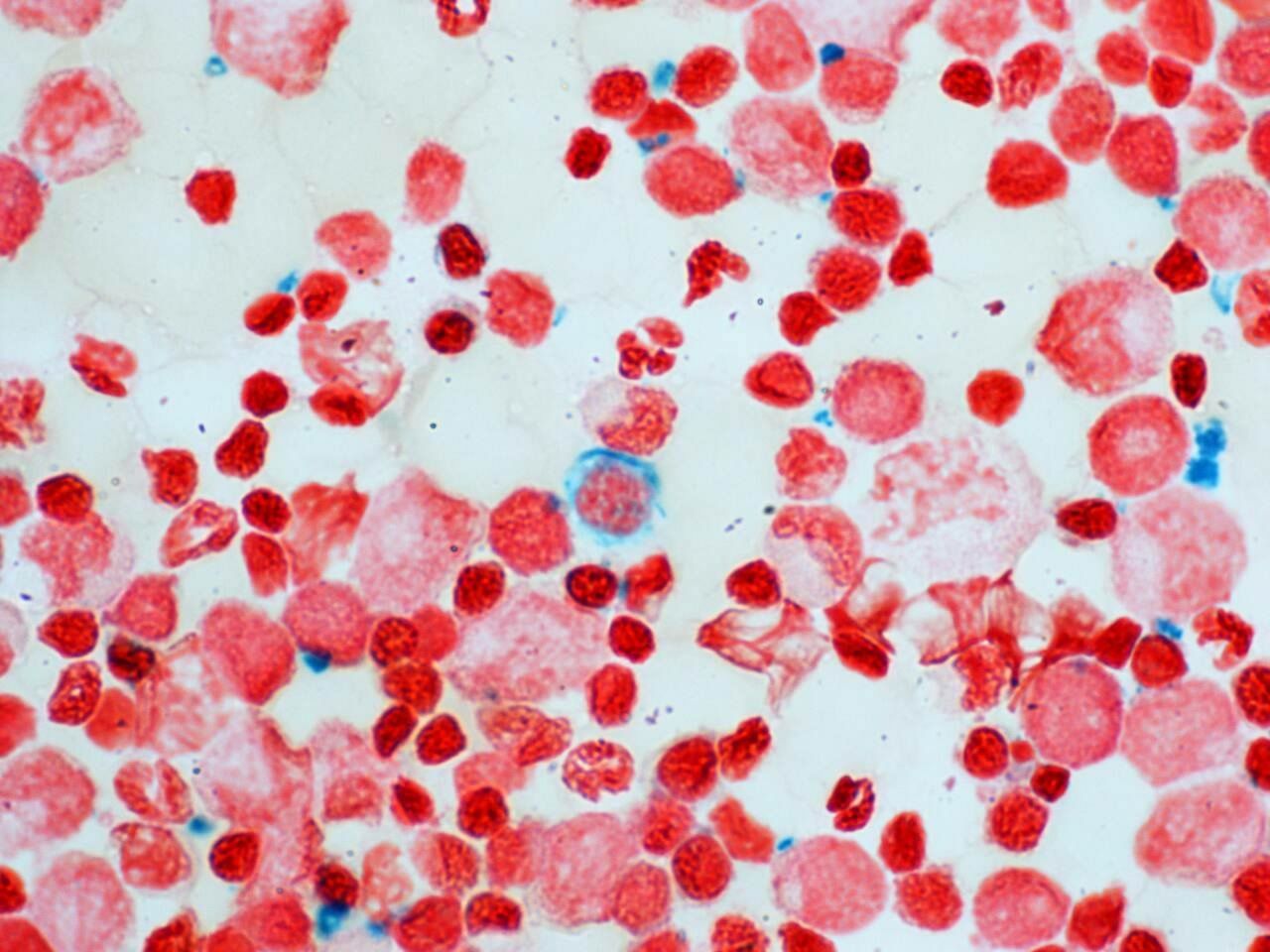
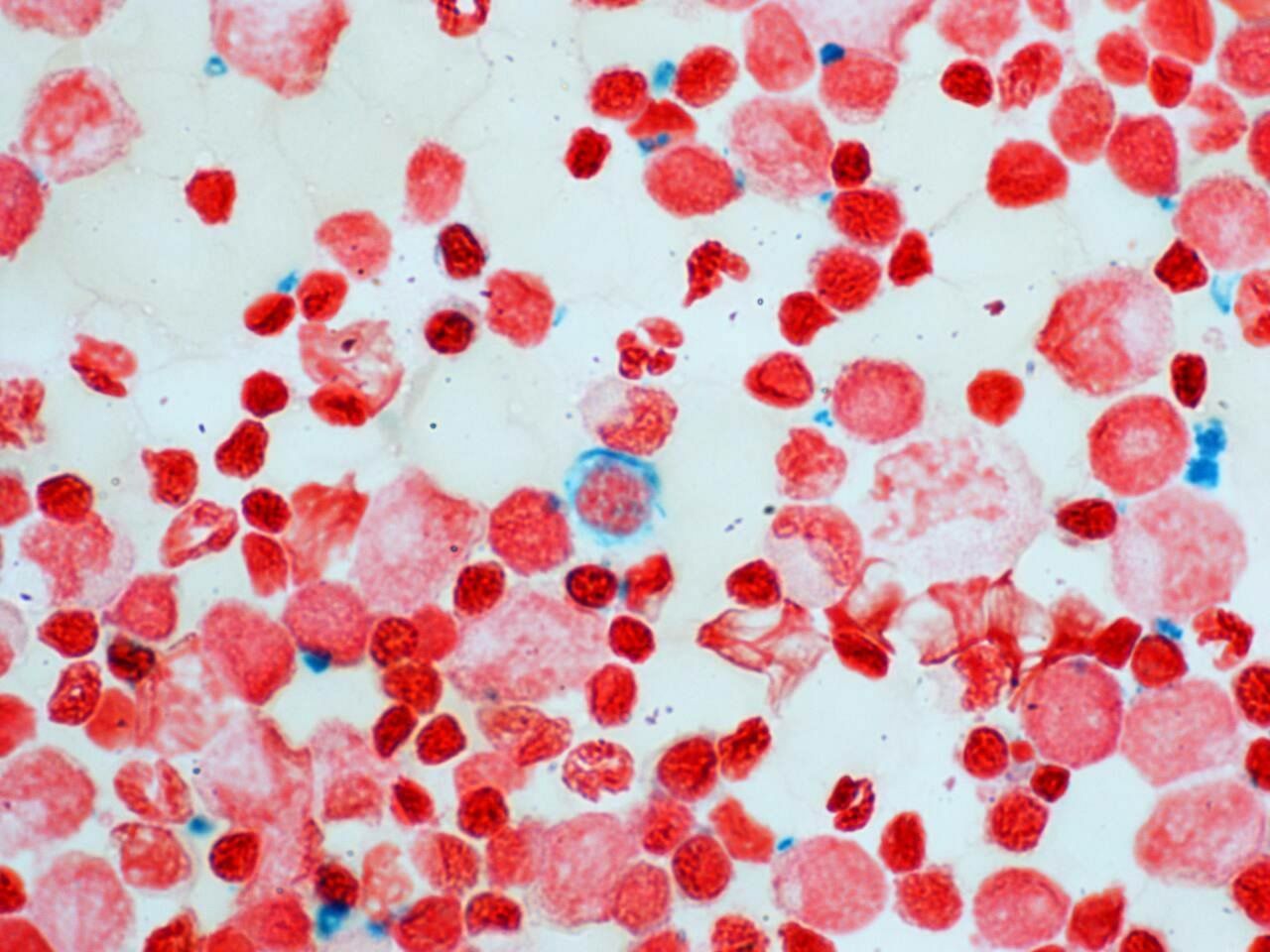
With transfusion support the nucleated red cell count gradually reduced, becoming absent on peripheral blood film. The child became transfusion dependent, with bone marrow aspirate demonstrating sideroblasts (figure 5), expanded erythropoiesis and iron overload. There was immunodeficiency (panhypogammaglobulinaemia) and the patient was given a clinical diagnosis of SIFD. There were no manifestations of periodic fever or significant developmental delay (mild gross motor only).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bone marrow aspirate demonstrating sideroblast.
Matched sibling bone marrow stem cell transplantation was undertaken at the age of 5 months. Myeloablative conditioning was performed with a reduced toxicity protocol of fludarabine, treosulfan and thiotepa, with alemtuzumab for serotherapy. The patient developed significant neurological complications soon after transplantation, including intractable seizures, with white matter and subsequently cystic changes on MR. Following admission to paediatric intensive care, he experienced a significant deterioration in his clinical status, including idiopathic pulmonary syndrome and ventilator dependence. Care was withdrawn with the consensus of the family, and the patient died peacefully on day 38 following transplant.
Mutation testing identified biallelic mutations in both affected children; TNRT1 c.608+1G>T and c.668T>C, and confirmed both parents as carriers.
Discussion
Fetal hydrops
Fetal hydrops describes a serious, and normally lethal fetal condition presenting with abnormal fluid accumulation in at least two distinct fetal compartments, including subcutaneous oedema, intra-abdominal fluid (ascites) and pleural and pericardial effusion.3 Association with placental oedema and thickening, and disturbance of amniotic volumes is well established, but not always identified on antenatal imaging.4 First presentation can often be at delivery, with poor condition at birth and very poor prognosis. The underlying pathophysiology is complex and remains poorly understood, but includes impaired lymphatic function, difficulty maintaining venous return, osmotic pressure and arterial pressures, and inadequate systemic perfusion leading to hypoxic injury and multiorgan failure.4 Mortality is approximately 60%–100%, depending on aetiology and case series. Prognosis is universally poor, with significant morbidity in survivors.5 Recognised aetiologies include congenital malformations, chromosomal abnormalities (eg, Turner’s syndrome), intrauterine infection (eg, cytomegalovirus), rhesus disease, severe anaemia (eg, thalassaemia) and maternal drug use (including prescription).4–6
Sideroblastic anaemia
SA describes disordered erythropoiesis, with erythroid precursor cells with perinuclear mitochondrial iron deposition visible in bone marrow.1 This gives the characteristic appearance of ‘ringed sideroblasts’, as seen on Perl’s staining (figure 1). The underlying pathology involves impaired mitochondrial function. Approximately 60% of cases of congenital sideroblastic anaemia (CSA) have an identifiable, underlying genetic mutation,7 with altered haem biosynthesis, iron-sulfur pathways or mitochondrial translation.1
SA with immunodeficiency, fevers and developmental delay
In 2015, a series of 11 children with SA of unknown cause and common phenotypic features were reviewed as part of an international collaboration.1 Patients were initially identified on the basis of SA (usually severe, microcytic, transfusion dependent) and B cell lymphopaenia and/or panhypogammaglobulinaemia alone.1 Subsequent review however found further clinical associations present in all cases; these included periodic fevers and developmental delay.1 Other clinical manifestations were identified across the cohort.1 These included cardiomyopathy, renal abnormalities (nephrocalcinosis/renal tubular dysfunction/Fanconi syndrome/aminoaciduria), retinitis pigmentosa, hepatosplenomegaly and cutaneous changes (ichthyotic skin changes, hypopigmentation, erythematous eruptions).1 Presentation is mostly common in the first 3 months of life (~60% of cases), and within the cohort described above all had presented by 7 months of age,1 with diagnosis of SA preceding immunodeficiency in all cases.1
Treatment of SIFD is supportive in the first instance, with regular and frequent blood transfusions, treatment of subsequent iron overload with iron chelation therapy, and intermittent immunoglobulin therapy. Increasingly allogeneic bone marrow transplant is being undertaken as a curative intervention, aiming to restore normal immune function and erythropoiesis. Cessation of neurological and developmental decline has been described post-transplant, and improvement in function is an established phenomenon.
Following the identification of SIFD as a distinct if rare form of CSA, an autosomal recessive pattern of inheritance has been established,2 with biallelic mutations identified in TRNT1, a gene encoding an enzyme involved in the addition of a conserved nucleotide triplet (CCA) to tRNA, affecting mitochondrial function. Partial loss of function can result in profound mitochondrial and metabolic dysfunction, and the clinical symptoms described above.
To our knowledge, this is the first case where SIFD has been described as the underlying cause of severe, chronic antenatal anaemia leading to fetal hydrops. PM genetic diagnosis was only possible following the development of SIFD in a subsequent sibling. Mutations in TRNT1 confirming SIFD should be tested for in cases of fetal hydrops presenting with severe anaemia and extramedullary haematopoiesis.
Take home messages
SIFD is rare, congenital anaemia identified initially through a global collaboration.
SIFD can present at birth with prominent extramedullary erythropoiesis and increased circulating nucleated red blood cells.
The SIFD phenotype can be extended to include fetal hydrops and severe congenital anaemia.
Footnotes
Handling editor Mary Frances McMullin.
Contributors All authors have seen the content of this submitted article, and are in agreement that it is correct in both scientific and clinical fact, and is original in its current form and content.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; internally peer reviewed.